
Tracing the Dynamical Genetic Diversity Changes of Russian Livni Pigs during the Last 50 Years with the Museum, Old, and Modern Samples

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
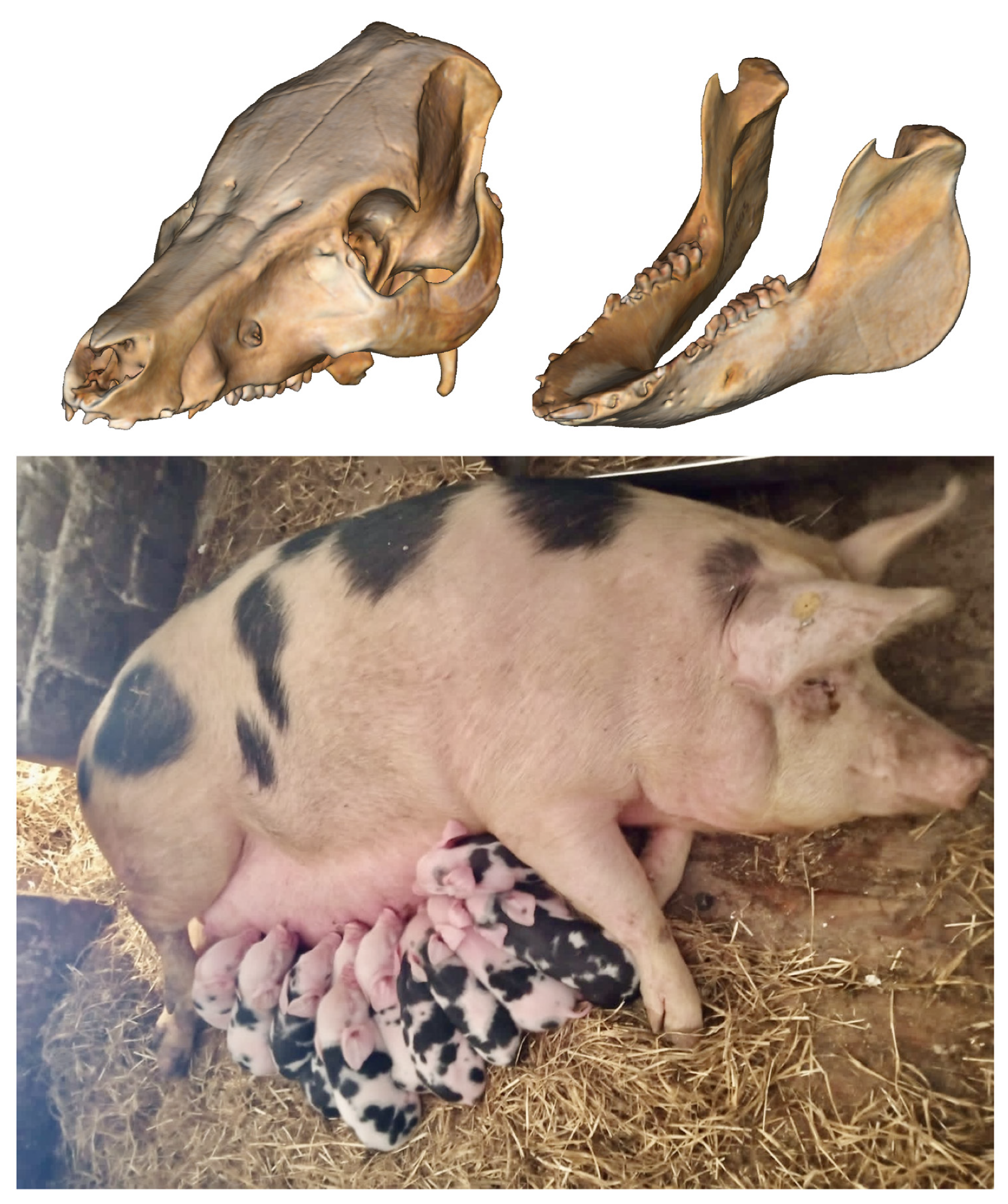
2.2. Sample Collection and DNA Extraction
2.3. SNP Genotyping and Quality Control
2.4. Genetic Diversity and Effective Population Size
2.5. Runs of Homozygosity (ROHs)
2.6. PCA, Neighbour-Net, and Admixture
2.7. TreeMix Analysis
2.8. D-Statistics and F-Statistics
3. Results
3.1. Genetic Diversity and Effective Population Size
3.2. Patterns of ROH Distribution
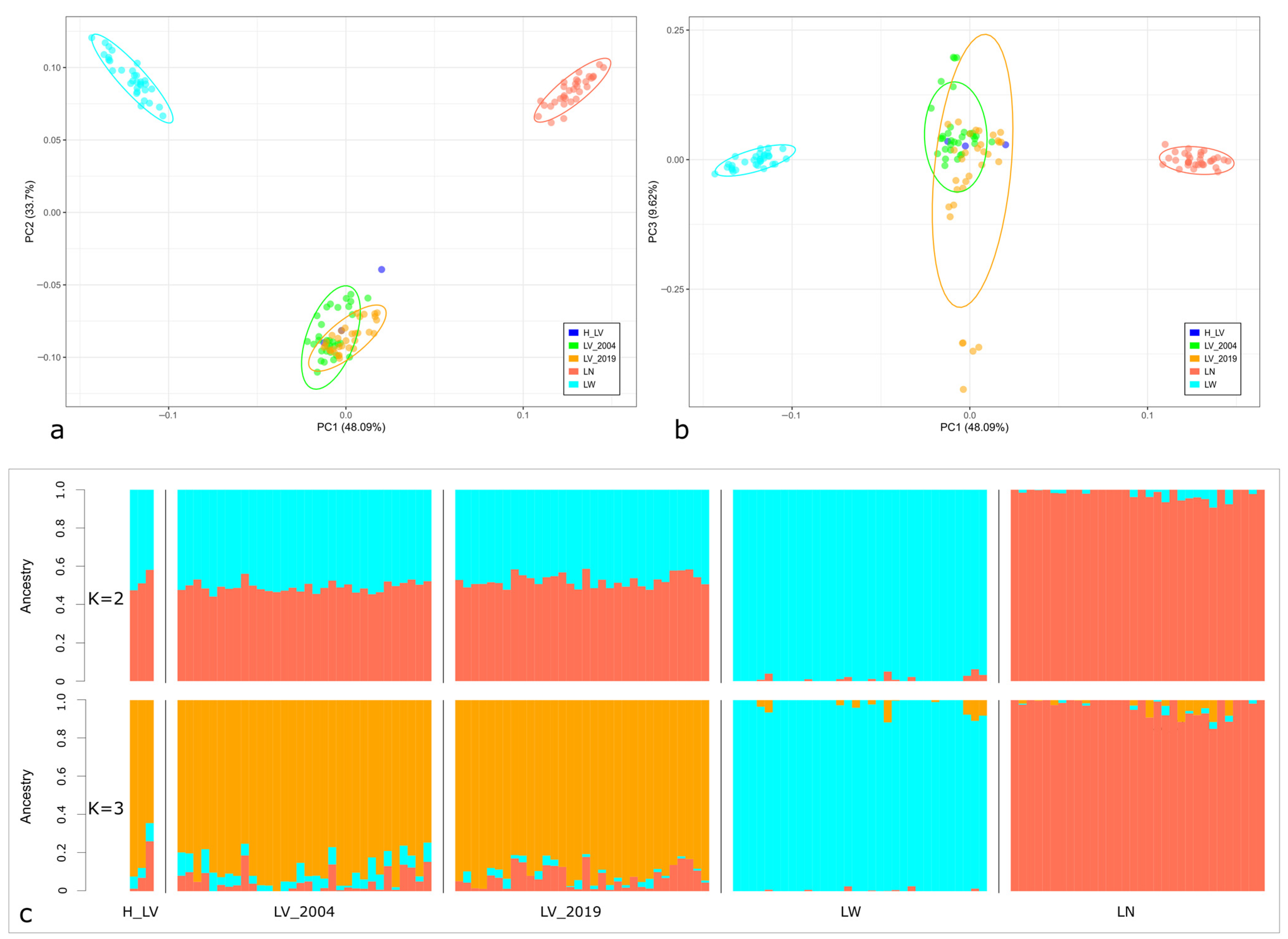
3.3. Genetic Differentiation and Admixture Patterns in the Studied Pig Populations
3.4. TreeMix Analysis
3.5. D-Statistics and F-Statistics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Comstock, R.E.; Robinson, H.F.; Harvey, P.H. A breeding procedure designed to make maximum use of both general and specific combining ability. Agron. J. 1949, 41, 360–367. [Google Scholar] [CrossRef]
- Sellier, P. The basis of crossbreeding in pigs. A review. Livest. Prod. Sci. 1976, 3, 203–226. [Google Scholar] [CrossRef]
- DAD-IS. Domestic Animal Diversity Information System (DAD-IS). Available online: http://www.fao.org/dad-is/ (accessed on 23 January 2024).
- Ernst, L.K.; Dmitriev, N.G.; Paronyan, I.A. Genetic Resources of Farm Animals in Russia and Neighboring Countries; All-Russian Research Institute of Animal Genetics and Breeding: St. Petersburg, Russia, 1994; p. 469. [Google Scholar]
- Traspov, A.; Deng, W.; Kostyunina, O.; Ji, J.; Shatokhin, K.; Lugovoy, S.; Yang, B.; Zinovieva, N.A.; Huang, L. Population structure and genome characterization of local pig breeds in Russia, Belorussia, Kazakhstan and Ukraine. Genet. Sel. Evol. 2016, 48, 16. [Google Scholar] [CrossRef]
- Deniskova, T.E.; Dotsev, A.V.; Selionova, M.I.; Kunz, E.; Medugorac, I.; Reyer, H.; Wimmers, K.; Barbato, M.; Traspov, A.A.; Brem, G.; et al. Population structure and genetic diversity of twenty-five Russian sheep breeds based on whole-genome genotyping. Genet. Sel. Evol. 2018, 50, 29. [Google Scholar] [CrossRef]
- Zinovieva, N.A.; Dotsev, A.V.; Sermyagin, A.A.; Deniskova, T.E.; Abdelmanova, A.S.; Kharzinova, V.R.; Sölkner, J.; Reyer, H.; Wimmers, K.; Brem, G. Selection signatures in two oldest Russian native cattle breeds revealed using high-density single nucleotide polymorphism analysis. PLoS ONE 2020, 15, e0242200. [Google Scholar] [CrossRef] [PubMed]
- Lukić, B.; Ferenčaković, M.; Šalamon, D.; Čačić, M.; Orehovački, V.; Iacolina, L.; Curik, I.; Cubric-Curik, V. Conservation Genomic Analysis of the Croatian Indigenous Black Slavonian and Turopolje Pig Breeds. Front. Genet. 2020, 11, 261. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Dotsev, A.V.; Romanov, M.N.; Stanishevskaya, O.I.; Gladyr, E.A.; Rodionov, A.N.; Vetokh, A.N.; Volkova, N.A.; Fedorova, E.S.; Gusev, I.V.; et al. Unveiling comparative genomic trajectories of selection in egg-type Russian White and meat-type White Cornish chickens. Biology 2021, 10, 876. [Google Scholar] [CrossRef] [PubMed]
- Zinovieva, N.A.; Sheiko, I.P.; Dotsev, A.V.; Sheiko, R.I.; Mikhailova, M.E.; Sermyagin, A.A.; Abdelmanova, A.S.; Kharzino-va, V.R.; Reyer, H.; Wimmers, K.; et al. Genome-wide SNP analysis clearly distinguished the Belarusian Red cattle from other European cattle breeds. Anim. Genet. 2021, 52, 720–724. [Google Scholar] [CrossRef]
- Volkopyalov, B.P.; Lus, J.J.; Shulzhenko, I.F. Breeds, Genetics and Breeding of Pigs; State Publishing House of Collective Farm and State Farm Literature: Moscow/Leningrad, Russia, 1934; p. 268. [Google Scholar]
- Bondarenko, A.F. Pig Breeds; State Publishing House of Collective Farm and State Farm Literature of the Ukrainian SSR: Lviv, Ukraine, 1940; p. 87. [Google Scholar]
- Lobanov, P.P. Agricultural Encyclopedia. Vol 3 (L–P), 3rd ed.; State Publishing House of Agricultural Literature: Moscow, Russia, 1953; p. 613. [Google Scholar]
- Gerasimov, V.I.; Berezovsky, M.D.; Nagaevich, V.M. The World Gene Pool of Pigs; Espada Publishing: Kharkiv, Ukraine, 2006; p. 520. [Google Scholar]
- Soldatov, A.P. Complete Catalog of Breeds of Agricultural Animals of Russia. Farm Animals; Eksmo-Press; Lik-Press: Moscow, Russia, 2001; p. 128. [Google Scholar]
- Tyapugin, S.E.; Butusov, D.V. (Eds.) Yearbook on Breeding Work in Pig Breeding in the Farms of the Russian Federation in 2020; The Publishing House of All-Russian Research Institute of Animal Breeding of the Russian Ministry for Agriculture: Lesnyye Polyany, Russia, 2021; 154p. [Google Scholar]
- Yang, B.; Cui, L.; Perez-Enciso, M.; Traspov, A.; Crooijmans, R.P.M.A.; Zinovieva, N.; Schook, L.B.; Archibald, A.; Gatphayak, K.; Knorr, C.; et al. Genome-wide SNP data unveils the globalization of domesticated pigs. Genet. Sel. Evol. 2017, 49, 71. [Google Scholar] [CrossRef]
- Sermyagin, A.A.; Dotsev, A.V.; Gladyr, E.A.; Traspov, A.A.; Deniskova, T.E.; Kostjunina, O.V.; Reyer, H.; Wimmers, K.; Barbato, M.; Paronyan, I.A.; et al. Whole-genome SNP analysis elucidates the genetic structure of Russian cattle and its relationship with Eurasian taurine breeds. Genet. Sel. Evol. 2018, 50, 37. [Google Scholar] [CrossRef]
- Gorssen, W.; Meyermans, R.; Buys, N.; Janssens, S. SNP genotypes reveal breed substructure, selection signatures and highly inbred regions in Piétrain pigs. Anim. Genet. 2020, 51, 32–42. [Google Scholar] [CrossRef]
- Deniskova, T.E.; Dotsev, A.V.; Selionova, M.I.; Reyer, H.; Sölkner, J.; Fornara, M.S.; Aybazov, A.-M.M.; Wimmers, K.; Brem, G.; Zinovieva, N.A. SNP-Based Genotyping Provides Insight into the West Asian Origin of Russian Local Goats. Front. Genet. 2021, 12, 1133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Oyelami, F.O.; Qadri, Q.R.; Sun, H.; Xu, Z.; Wang, Q.-s.; Pan, Y. Identifying the unique characteristics of the Chinese indigenous pig breeds in the Yangtze River Delta region for precise conservation. BMC Genom. 2021, 22, 151. [Google Scholar] [CrossRef]
- Rowe, K.C.; Singhal, S.; Macmanes, M.D.; Ayroles, J.F.; Morelli, T.L.; Rubidge, E.M.; Bi, K.; Moritz, C.C. Museum genomics: Low-cost and high-accuracy genetic data from historical specimens. Mol. Ecol. Resour. 2011, 11, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Billerman, S.M.; Walsh, J. Historical DNA as a tool to address key questions in avian biology and evolution: A review of methods, challenges, applications, and future directions. Mol. Ecol. Resour. 2019, 19, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Zinovieva, N.A.; Sermyagin, A.A.; Dotsev, A.V.; Boronetslaya, O.I.; Petrikeeva, L.V.; Abdelmanova, A.S.; Brem, G. Animal genetic resources: Developing the research of allele pool of Russian cattle breeds—Minireview. Agric. Biol. 2019, 54, 631–641. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.V.; Mishina, A.I.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.I.; Petrikeeva, L.V.; Chinarov, R.Y.; Brem, G.; et al. Genetic diversity of the historical and modern populations of Russian cattle breeds revealed by microsatellite analysis. Genes 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Raxworthy, C.J.; Smith, B.T. Mining museums for historical DNA: Advances and challenges in museomics. Trends Ecol. Evol. 2021, 36, 1049–1060. [Google Scholar] [CrossRef]
- McHugo, G.P.; Dover, M.J.; MacHugh, D.E. Unlocking the origins and biology of domestic animals using ancient DNA and paleogenomics. BMC Biol. 2019, 17, 98. [Google Scholar] [CrossRef]
- Buggiotti, L.; Yurchenko, A.A.; Yudin, N.S.; Van der Jagt, C.J.; Vorobieva, N.V.; Kusliy, M.A.; Vasiliev, S.K.; Rodionov, A.N.; Boronetskaya, O.I.; Zinovieva, N.A.; et al. Demographic history, adaptation, and NRAP convergent evolution in the world northernmost cattle from Siberia. Mol. Biol. Evol. 2021, 38, msab078. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.V.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.I.; Chinarov, R.Y.; Lutshikhina, E.M.; Sölkner, J.; Brem, G.; et al. Comparative study of the genetic diversity of local steppe cattle breeds from Russia, Kazakhstan and Kyrgyzstan by microsatellite analysis of museum and modern samples. Diversity 2021, 13, 351. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Meyer, M.; Krause, J.; Kircher, M.; Paabo, S. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010, 38, e87. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 20 October 2023).
- Fan, J.B.; Oliphant, A.; Shen, R.; Kermani, B.G.; Garcia, F.; Gunderson, K.L.; Hansen, M.; Steemers, F.; Butler, S.L.; Deloukas, P.; et al. Highly parallel SNP genotyping. Cold Spring Harb. Symp. Quant. Biol. 2003, 68, 69–78. [Google Scholar] [CrossRef]
- Nei, M. Estimation of average heterozygosity and genetic distance from small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodohl, P.A. diveRsity: An R package for the estimation of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Corbin, L.J.; Liu, A.Y.; Bishop, S.C.; Woolliams, J.A. Estimation of historical effective population size using linkage disequilibria with marker data. J. Anim. Breed. Genet. 2012, 129, 257–270. [Google Scholar] [CrossRef]
- Sved, J.A.; Feldman, M.W. Correlation and probability methods for one and two loci. Theor. Popul. Biol. 1973, 4, 129–132. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsam, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2014, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Biscarini, F.; Paolo Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. R Package Version 0.9.5. Available online: https://cran.r-project.org/web/packages/detectRUNS/index.html (accessed on 8 December 2023).
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef] [PubMed]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homo-zygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, M.; Capomaccio, S.; Vajana, E.; Bomba, L.; Garcia, J.F.; Ajmone-Marsan, P.; Colli, L. BITE: An R package for biodiversity analyses. bioRxiv 2017, 181610. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Pritchard, J.K. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Fitak, R.R. OptM: Estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 2021, 6, bpab017. [Google Scholar] [CrossRef]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.; et al. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef]
- Patterson, N.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient Admixture in Human History. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef]
- Petr, M.; Vernot, B.; Kelso, J. Admixr—R package for reproducible analyses using ADMIXTOOLS. Bioinformatics 2019, 35, 3194–3195. [Google Scholar] [CrossRef] [PubMed]
- Scherf, B.D.; Pilling, D. The Second Report on the State of the World’s Animal Genetic Resources for Food and Agriculture; FAO Commission on Genetic Resources for Food and Agriculture Assessments: Rome, Italy, 2015; Available online: http://www.fao.org/3/a-i4787e/index.html (accessed on 28 January 2024).
- Chernukha, I.; Abdelmanova, A.; Kotenkova, E.; Kharzinova, V.; Zinovieva, N.A. Assessing Genetic Diversity and Searching for Selection Signatures by Comparison between the Indigenous Livni and Duroc Breeds in Local Livestock of the Central Region of Russia. Diversity 2022, 14, 859. [Google Scholar] [CrossRef]
- Bâlteanu, V.A.; Cardoso, T.F.; Amills, M.; Egerszegi, I.; Anton, I.; Beja-Pereira, A.; Zsolnai, A. The footprint of recent and strong demographic decline in the genomes of Mangalitza pigs. Animal 2019, 13, 2440–2446. [Google Scholar] [CrossRef]
- Zorc, M.; Škorput, D.; Gvozdanović, K.; Margeta, P.; Karolyi, D.; Luković, Z.; Salajpal, K.; Savić, R.; Muñoz, M.; Bovo, S.; et al. Genetic diversity and population structure of six autochthonous pig breeds from Croatia, Serbia, and Slovenia. Genet. Sel. Evol. 2022, 54, 30. [Google Scholar] [CrossRef] [PubMed]
- Mikhailova, O.A. Improving the meat productivity of Livenskaya breed of pigs under the conditions of peasant farms. Bull. Agrar. Sci. 2019, 4, 65–72. [Google Scholar] [CrossRef]
- Ewart, K.M.; Johnson, R.N.; Ogden, R.; Joseph, L.; Frankham, G.J.; Lo, N. Museum specimens provide reliable SNP data for population genomic analysis of a widely distributed but threatened cockatoo species. Mol. Ecol. Resour. 2019, 19, 1578–1592. [Google Scholar] [CrossRef]
- Byerly, P.A.; Chesser, R.T.; Fleischer, R.C.; McInerney, N.; Przelomska, N.A.S.; Leberg, P.L. Museum Genomics Provide Evidence for Persistent Genetic Differentiation in a Threatened Seabird Species in the Western Atlantic. Integr. Comp. Biol. 2022, 62, 1838–1848. [Google Scholar] [CrossRef]
- Parejo, M.; Wragg, D.; Henriques, D.; Charrière, J.D.; Estonba, A. Digging into the Genomic Past of Swiss Honey Bees by Whole-Genome Sequencing Museum Specimens. Genome Biol. Evol. 2020, 12, 2535–2551. [Google Scholar] [CrossRef]
- Kvist, L.; Honka, J.; Salazar, D.; Kirkinen, T.; Hemmann, K. Memories, museum artefacts and excavations in resolving the history of maternal lineages in the Finnhorse. Anim. Genet. 2022, 53, 821–828. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 Population | 2 n | 3 HO (7 M ± 8 SE) | 4 UHE (M ± SE) | 5 AR (M ± SE) | 6 UFIS [CI 95%] |
|---|---|---|---|---|---|
| H_LV | 3 | 0.383 ± 0.002 | 0.392 ± 0.001 | 1.775 ± 0.003 | 0.022 [0.016; 0.028] |
| LV_2004 | 32 | 0.409 ± 0.001 | 0.404 ± 0.001 | 1.798 ± 0.001 | −0.011 [−0.013; −0.009] |
| LV_2019 | 32 | 0.405 ± 0.001 | 0.401 ± 0.001 | 1.793 ± 0.001 | −0.009 [−0.011; −0.007] |
| LN | 32 | 0.366 ± 0.001 | 0.351 ± 0.001 | 1.703 ± 0.002 | −0.038 [−0.04; −0.036] |
| LW | 32 | 0.345 ± 0.001 | 0.333 ± 0.001 | 1.668 ± 0.002 | −0.033 [−0.035; −0.031] |
| 1 Population | 2 n | 3 ROH Length, Mb | 4 ROH Number | 5 FROH | ||||
|---|---|---|---|---|---|---|---|---|
| 6 M ± 7 SE | 8 MIN | 9 MAX | M ± SE | MIN | MAX | M ± SE | ||
| H_LV | 3 | 313.51 ± 96.88 | 119.77 | 412.15 | 214.67 ± 71.55 | 73 | 303 | 0.128 ± 0.04 |
| LV_2004 | 32 | 431.20 ± 12.58 | 320.43 | 589.78 | 216.06 ± 3.51 | 189 | 257 | 0.176 ± 0.005 |
| LV_2019 | 32 | 451.57 ± 13.16 | 330.77 | 659.71 | 213.62 ± 2.81 | 188 | 240 | 0.184 ± 0.005 |
| LN | 32 | 686.13 ± 15.73 | 517.98 | 851.82 | 249.53 ± 2.66 | 220 | 283 | 0.280 ± 0.006 |
| LW | 32 | 792.78 ± 14.73 | 670.87 | 1059.59 | 285.59 ± 2.17 | 266 | 314 | 0.324 ± 0.006 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelmanova, A.A.; Deniskova, T.E.; Kharzinova, V.R.; Chinarov, R.Y.; Boronetskaya, O.I.; Sölkner, J.; Brem, G.; Ai, H.; Huang, L.; Trukhachev, V.I.; et al. Tracing the Dynamical Genetic Diversity Changes of Russian Livni Pigs during the Last 50 Years with the Museum, Old, and Modern Samples. Animals 2024, 14, 1629. https://doi.org/10.3390/ani14111629
Abdelmanova AA, Deniskova TE, Kharzinova VR, Chinarov RY, Boronetskaya OI, Sölkner J, Brem G, Ai H, Huang L, Trukhachev VI, et al. Tracing the Dynamical Genetic Diversity Changes of Russian Livni Pigs during the Last 50 Years with the Museum, Old, and Modern Samples. Animals. 2024; 14(11):1629. https://doi.org/10.3390/ani14111629
Chicago/Turabian StyleAbdelmanova, Alexandra A., Tatiana E. Deniskova, Veronika R. Kharzinova, Roman Yu Chinarov, Oksana I. Boronetskaya, Johann Sölkner, Gottfried Brem, Huashui Ai, Lusheng Huang, Vladimir I. Trukhachev, and et al. 2024. "Tracing the Dynamical Genetic Diversity Changes of Russian Livni Pigs during the Last 50 Years with the Museum, Old, and Modern Samples" Animals 14, no. 11: 1629. https://doi.org/10.3390/ani14111629