

A Comparative Full-Length Transcriptome Analysis Using Oxford Nanopore Technologies (ONT) in Four Tissues of Bovine Origin
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. RNA Extraction and cDNA Library Construction
2.3. ONT-Based Long-Read Processing
2.4. Identification of Fusion Transcripts
2.5. Structural Analyses
2.6. Prediction of Transcription Factors (TFs)
2.7. Gene Functional Annotation
2.8. Quantification of Gene/Transcript Expression Levels and Differential Expression Analyses
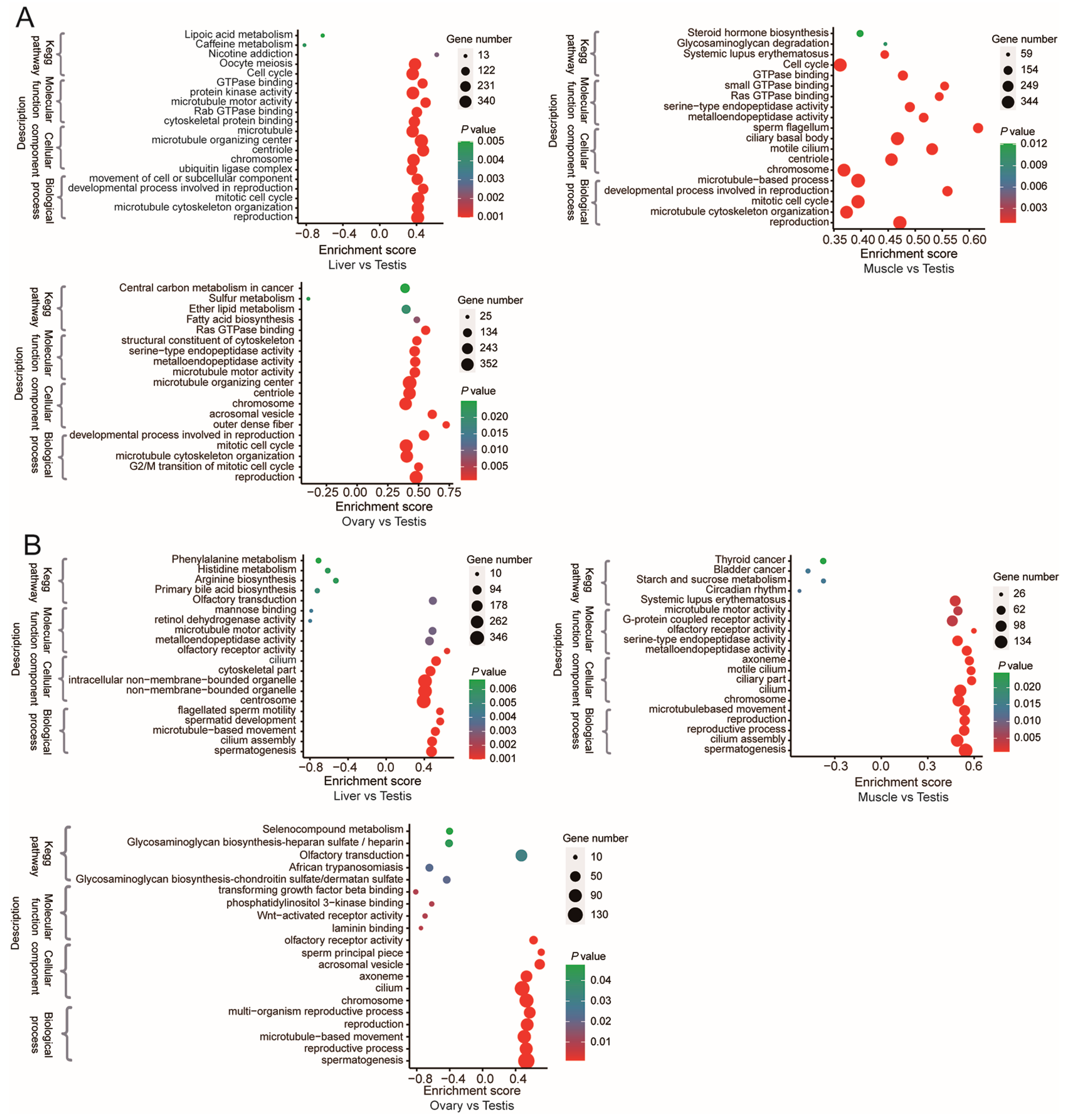
2.9. Functional Enrichment Analyses
2.10. Protein–Protein Interaction (PPI) Analyses
2.11. TF–Gene Interaction Network Analyses Were Performed Using the Network Analyst Tool and the JASPAR Database
3. Results
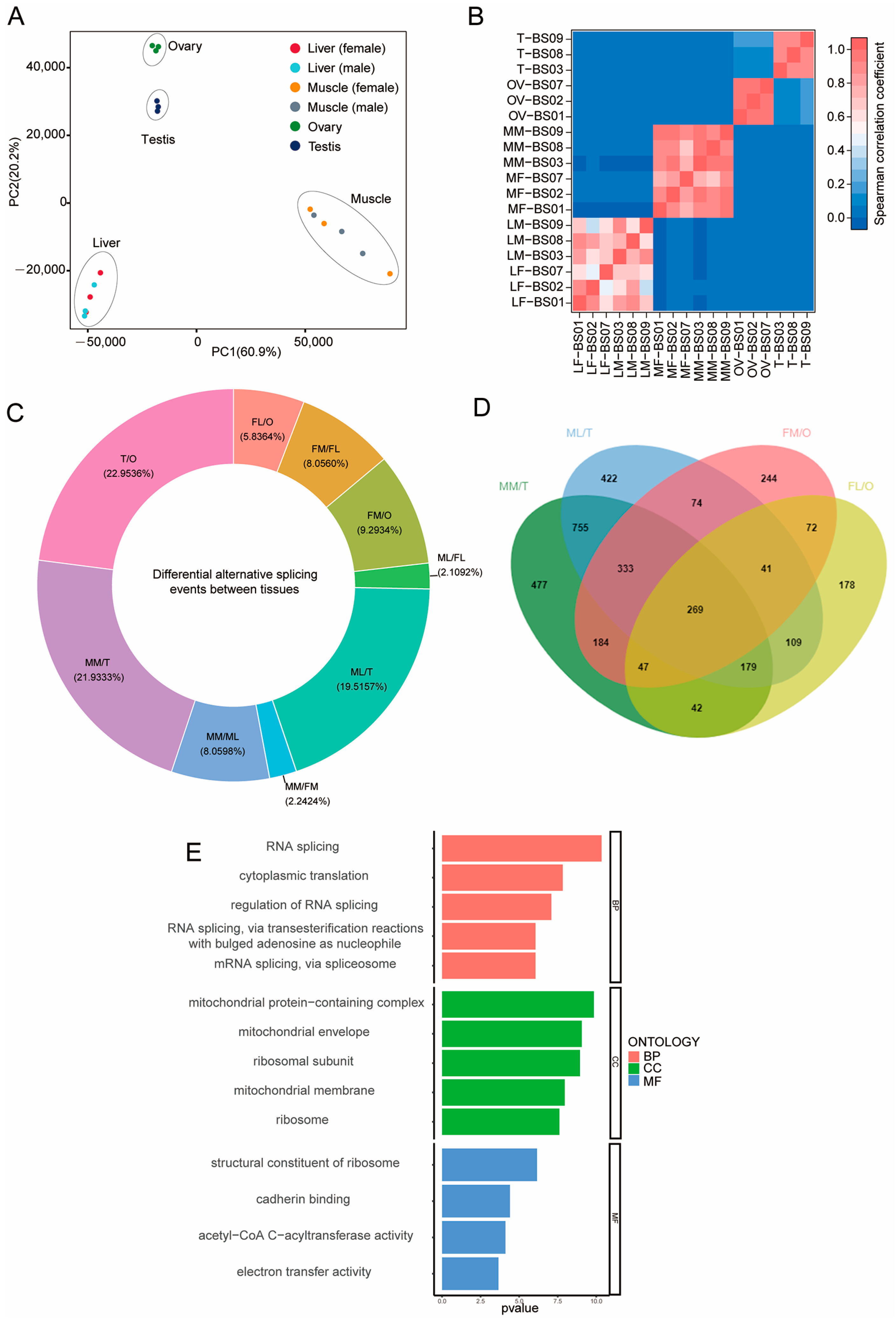
3.1. Alternative Splicing Structural Analyses of Bovine Tissues
3.2. APA Analyses of Bovine Tissues
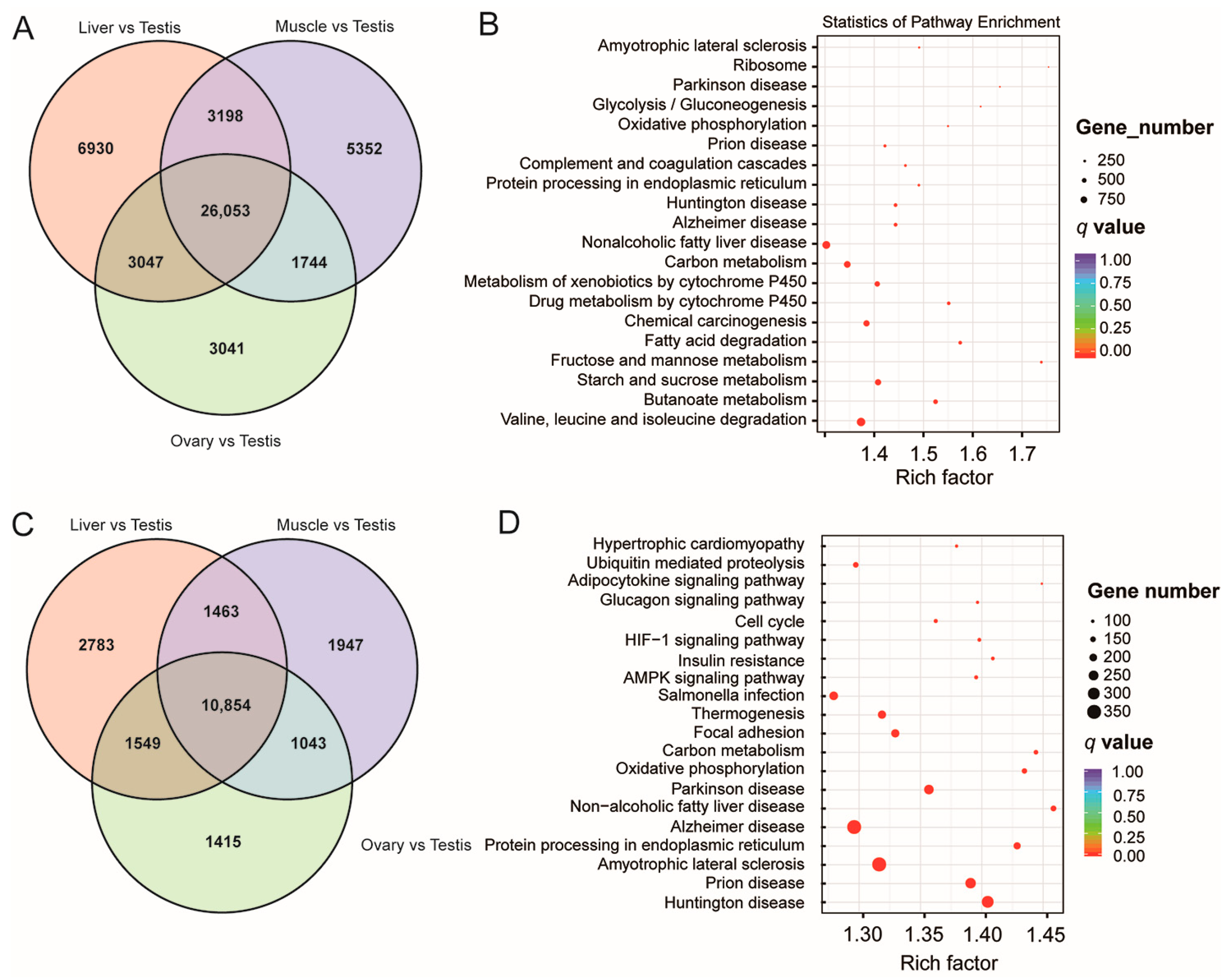
3.3. DETs and DEGs among the Various Tissues Indicated the Complexity of Testicular Expression
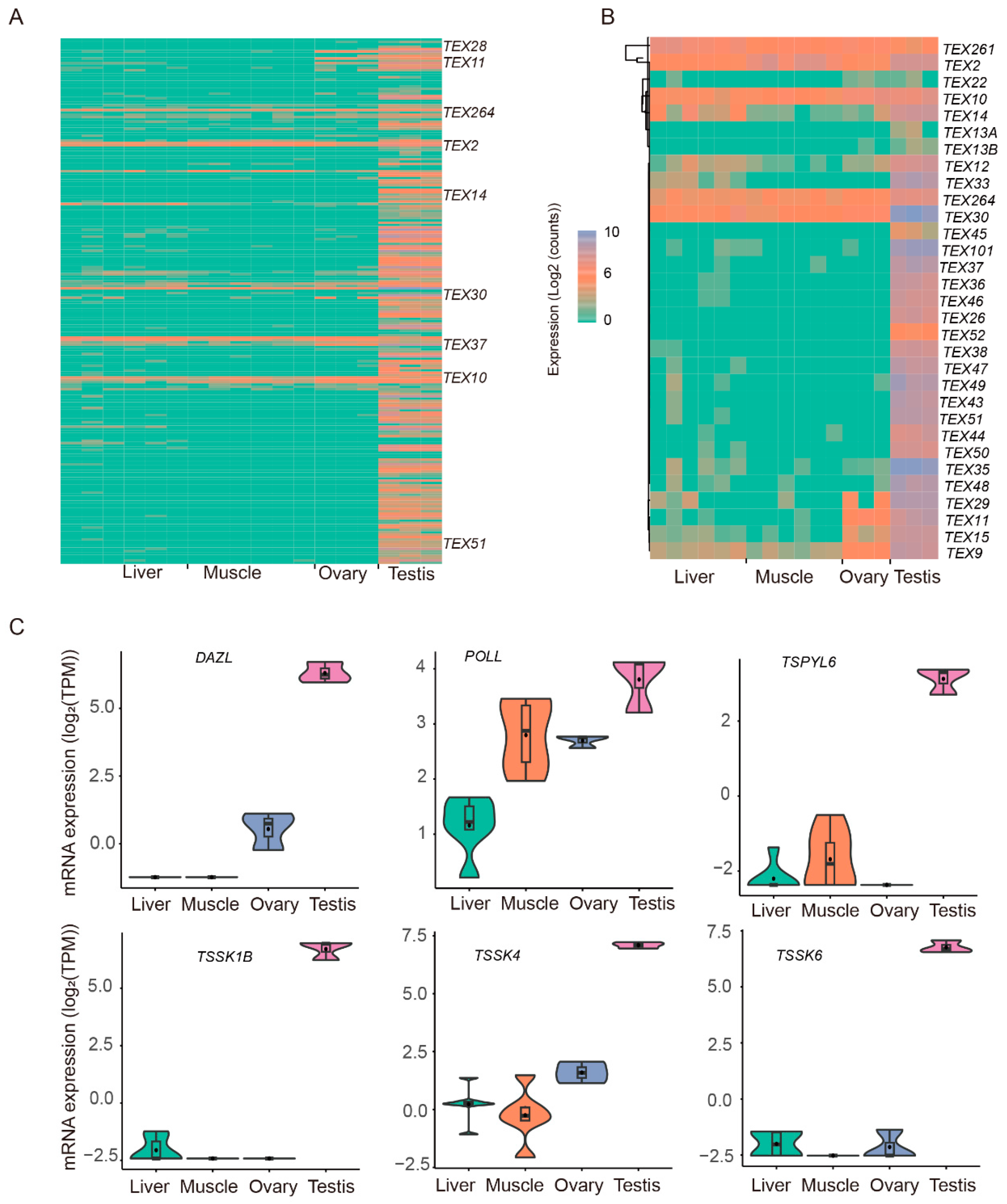
3.4. Testicular-Specific High-Expression Genes/Transcripts and Their Functional Annotation
3.5. Expression of TEST-Specific Genes
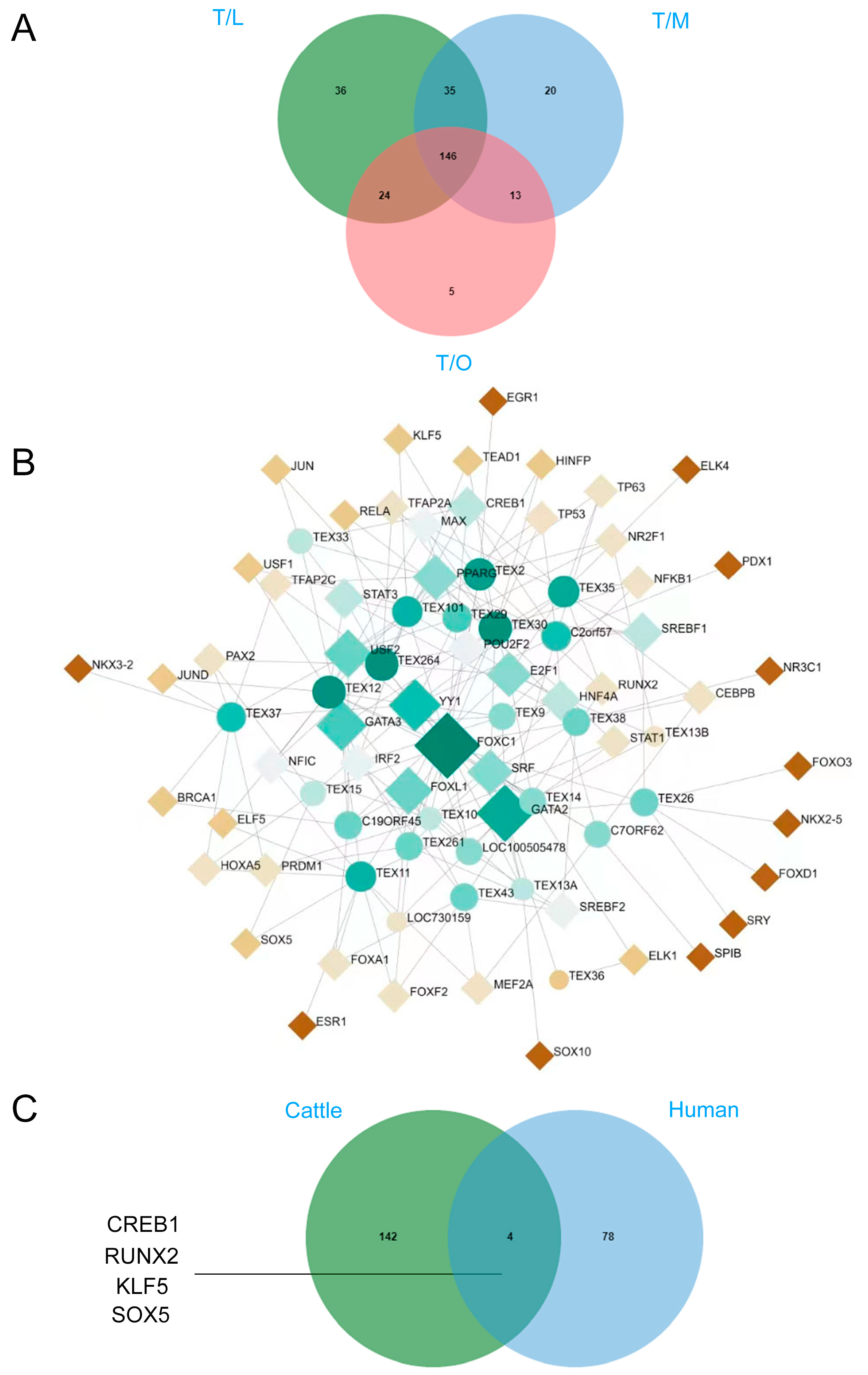
3.6. Identification of TEST Specifically Expressed Transcription Factors
4. Discussion
4.1. TESTs Have the Greatest Number of Differential Alternative Splicing Events and Genes with APA Events
4.2. Exploring Testicular-Specific Expression Genes by Comparing the Gene Expression of Four Tissues in Bovines
4.3. Identification of TEX Gene Transcripts and Prediction of Their Testicular-Specific Transcription Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, T.P.L.; Bickhart, D.M.; Boichard, D.; Chamberlain, A.J.; Djikeng, A.; Jiang, Y.; Low, W.Y.; Pausch, H.; Demyda-Peyrás, S.; Prendergast, J.; et al. The Bovine Pangenome Consortium: Democratizing production and accessibility of genome assemblies for global cattle breeds and other bovine species. Genome Biol. 2023, 24, 139. [Google Scholar] [CrossRef]
- Lecluze, E.; Jégou, B.; Rolland, A.D.; Chalmel, F. New transcriptomic tools to understand testis development and functions. Mol. Cell. Endocrinol. 2018, 468, 47–59. [Google Scholar] [CrossRef]
- Naro, C.; Cesari, E.; Sette, C. Splicing regulation in brain and testis: Common themes for highly specialized organs. Cell Cycle 2021, 20, 480–489. [Google Scholar] [CrossRef]
- Bellil, H.; Ghieh, F.; Hermel, E.; Mandon-Pepin, B.; Vialard, F. Human testis-expressed (TEX) genes: A review focused on spermatogenesis and male fertility. Basic Clin. Androl. 2021, 31, 9. [Google Scholar] [CrossRef]
- Guo, J.; Grow, E.J.; Mlcochova, H.; Maher, G.J.; Lindskog, C.; Nie, X.; Guo, Y.; Takei, Y.; Yun, J.; Cai, L.; et al. The adult human testis transcriptional cell atlas. Cell Res. 2018, 28, 1141–1157. [Google Scholar] [CrossRef]
- Wright, E.; Hargrave, M.R.; Christiansen, J.; Cooper, L.; Kun, J.; Evans, T.; Gangadharan, U.; Greenfield, A.; Koopman, P. The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat. Genet. 1995, 9, 15–20. [Google Scholar] [CrossRef]
- Menke, D.B.; Mutter, G.L.; Page, D.C. Expression of DAZ, an azoospermia factor candidate, in human spermatogonia. Am. J. Hum. Genet. 1997, 60, 237–241. [Google Scholar]
- Balhorn, R. The protamine family of sperm nuclear proteins. Genome Biol. 2007, 8, 227. [Google Scholar] [CrossRef]
- Aoki, V.W.; Emery, B.R.; Liu, L.; Carrell, D.T. Protamine levels vary between individual sperm cells of infertile human males and correlate with viability and DNA integrity. J. Androl. 2006, 27, 890–898. [Google Scholar] [CrossRef]
- Wang, X.; Li, H.; Fu, G.; Wang, Y.; Du, S.; Yu, L.; Wei, Y.; Chen, S. Testis-specific serine/threonine protein kinase 4 (Tssk4) phosphorylates Odf2 at Ser-76. Sci. Rep. 2016, 6, 22861. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Deamer, D.; Akeson, M.; Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef]
- Li, Y.; Fang, C.; Fu, Y.; Hu, A.; Li, C.; Zou, C.; Li, X.; Zhao, S.; Zhang, C.; Li, C. A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing. DNA Res. 2018, 25, 421–437. [Google Scholar] [CrossRef]
- Li, D.; Zhong, C.; Sun, Y.; Kang, L.; Jiang, Y. Identification of genes involved in chicken follicle selection by ONT sequencing on granulosa cells. Front. Genet. 2022, 13, 1090603. [Google Scholar] [CrossRef]
- Lamb, H.J.; Ross, E.M.; Nguyen, L.T.; Lyons, R.E.; Moore, S.S.; Hayes, B.J. Characterization of the poll allele in Brahman cattle using long-read Oxford Nanopore sequencing. J. Anim. Sci. 2020, 98, skaa127. [Google Scholar] [CrossRef]
- Halstead, M.M.; Islas-Trejo, A.; Goszczynski, D.E.; Medrano, J.F.; Zhou, H.; Ross, P.J. Large-Scale Multiplexing Permits Full-Length Transcriptome Annotation of 32 Bovine Tissues from a Single Nanopore Flow Cell. Front. Genet. 2021, 12, 664260. [Google Scholar] [CrossRef]
- Sun, L.Y.; Qu, K.X.; Liu, Y.K.; Ma, X.H.; Chen, L.B.; Zhang, J.C.; Huang, B.Z.; Lei, C.Z. Assessing genomic diversity and selective pressures in Bashan cattle by whole-genome sequencing data. Anim. Biotechnol. 2023, 34, 835–846. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Cai, Y.W.; He, F.C. Integrated NR Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 5, 2699. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 1, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analyses of protein functions and evolution. Nucleic Acids Res. 2000, 1, 33–36. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Raoet, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 2, R7. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 1, 27–30. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analyses for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Hu, H.; Miao, Y.R.; Jia, L.H.; Yu, Q.Y.; Zhang, Q.; Guo, A.Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- MacDonald, C.C.; Redondo, J.L. Reexamining the polyadenylation signal: Were we wrong about AAUAAA? Mol. Cell. Endocrinol. 2002, 190, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Graber, J.H. Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip. Rev. RNA 2012, 3, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. TFSyntax: A database of transcription factors binding syntax in mammalian genomes. Nucleic Acids Res. 2023, 51, D306–D314. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.M.; Albu, M.; Chen, X.T.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, A.; Cole, J.B.; VanRaden, P.M.; Wiggans, G.R.; Ruiz-López, F.J.; Van Tassell, C.P. Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc. Natl. Acad. Sci. USA 2016, 113, E3995–E4004. [Google Scholar] [CrossRef] [PubMed]
- Thompson-Crispi, K.; Atalla, H.; Miglior, F.; Mallard, B.A. Bovine mastitis: Frontiers in immunogenetics. Front. Immunol. 2014, 5, 493. [Google Scholar] [CrossRef] [PubMed]
- Saatchi, M.; Schnabel, R.D.; Rolf, M.M.; Taylor, J.F.; Garrick, D.J. Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet. Sel. Evol. 2012, 44, 38. [Google Scholar] [CrossRef] [PubMed]
- Meredith, B.K.; Kearney, F.J.; Finlay, E.K.; Bradley, D.G.; Fahey, A.G.; Berry, D.P.; Lynn, D.J. Genome-wide associations for milk production and somatic cell score in Holstein-Friesian cattle in Ireland. BMC Genet. 2012, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Lervik, S.; Kristoffersen, A.B.; Conley, L.N.; Oskam, I.C.; Hedegaard, J.; Ropstad, E.; Olsaker, I. Gene expression during testis development in Duroc boars. Animal 2015, 9, 1832–1842. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, S.; Duan, Q.; Lou, M.; Ling, Y. Comprehensive analyses of 435 goat transcriptomes provides insight into male reproduction. Int. J. Biol. Macromol. 2024, 255, 127942. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhang, X.; Yan, J.; Guo, J.; Zhang, F.; Zhu, K.; Liu, S.; Sun, Y.; Shen, W.; Wang, J. Transcriptional Specificity Analyses of testis and Epididymis Tissues in Donkey. Genes 2022, 13, 2339. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Lee, J.Y.; Pan, Z.; Jiang, B.; Tian, B. Progressive lengthening of 3′ untranslated regions of mRNAs by APA during mouse embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 7028–7033. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Brockman, J.M.; Dass, B.; Hutchins, L.N.; Singh, P.; McCarrey, J.R.; MacDonald, C.C.; Graber, J.H. Systematic variation in mRNA 3′-processing signals during mouse spermatogenesis. Nucleic Acids Res. 2007, 35, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.; Grellscheid, S.N.; Ehrmann, I.; Dalgliesh, C.; Danilenko, M.; Paronetto, M.P.; Pedrotti, S.; Grellscheid, D.; Dixon, R.J.; Sette, C.; et al. The splicing landscape is globally reprogrammed during male meiosis. Nucleic Acids Res. 2013, 41, 10170–10184. [Google Scholar] [CrossRef] [PubMed]
- Zagore, L.L.; Grabinski, S.E.; Sweet, T.J.; Hannigan, M.M.; Sramkoski, R.M.; Li, Q.; Licatalosi, D.D. RNA Binding Protein Ptbp2 Is Essential for Male Germ Cell Development. Mol. Cell. Biol. 2015, 35, 4030–4042. [Google Scholar] [CrossRef]
- MacDonald, C.C. Tissue-specific mechanisms of APA: Testis, brain, and beyond (2018 update). Wiley Interdiscip. Rev. RNA 2019, 10, e1526. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.; Zheng, X.; Manske, G.L.; Vargo, A.; Shami, A.N.; Li, J.Z.; Hammoud, S.S. Decoding the Spermatogenesis Program: New Insights from Transcriptomic Analyses. Annu. Rev. Genet. 2022, 56, 339–368. [Google Scholar] [CrossRef]
- Schultz, N.; Hamra, F.K.; Garbers, D.L. A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc. Natl. Acad. Sci. USA 2003, 100, 12201–12206. [Google Scholar] [CrossRef]
- Lu, Y.; Oura, S.; Matsumura, T.; Oji, A.; Sakurai, N.; Fujihara, Y.; Shimada, K.; Miyata, H.; Tobita, T.; Noda, T.; et al. CRISPR/Cas9-mediated genome editing reveals 30 testis-enriched genes dispensable for male fertility in mice. Biol. Reprod. 2019, 101, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.J.; McCarrey, J.R.; Yang, F.; Page, D.C. An abundance of X-linked genes expressed in spermatogonia. Nat. Genet. 2001, 27, 422–426. [Google Scholar] [CrossRef]
- Sieper, M.H.; Gaikwad, A.S.; Fros, M.; Weber, P.; Di Persio, S.; Oud, M.S.; Kliesch, S.; Neuhaus, N.; Stallmeyer, B.; Tüttelmann, F.; et al. Scrutinizing the human TEX genes in the context of human male infertility. Andrology 2023, 12, 570–584. [Google Scholar] [CrossRef]
- Gholamitabar Tabari, M.; Jorsaraei, S.G.A.; Ghasemzadeh-Hasankolaei, M.; Ahmadi, A.A.; Mahdinezhad Gorji, N. Evaluation of specific germ cell genes expression in mouse embryonic stem cell-derived germ cell like cells treated with bone morphogenetic protein 4 in vitro. Int. J. Reprod. Biomed. 2018, 16, 507–518. [Google Scholar] [CrossRef]
- Otowa, T.; Yoshida, E.; Sugaya, N.; Yasuda, S.; Nishimura, Y.; Inoue, K.; Tochigi, M.; Umekage, T.; Miyagawa, T.; Nishida, N.; et al. Genome-wide association study of panic disorder in the Japanese population. J. Hum. Genet. 2009, 54, 122–126. [Google Scholar] [CrossRef]
- Lee, K.W.; Ching, S.M.; Devaraj, N.K.; Hoo, F.K. Genetic polymorphisms in neuroendocrine disorder-related candidate genes associated with pre-pregnancy obesity in gestational diabetes mellitus patients by using a stratification approach. Ann. Transl. Med. 2020, 8, 1060. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Lamb, D.J. The biology of infertility: Research advances and clinical challenges. Nat. Med. 2008, 14, 1197–1213. [Google Scholar] [CrossRef]
- Greenbaum, M.P.; Yan, W.; Wu, M.H.; Lin, Y.N.; Agno, J.E.; Sharma, M.; Braun, R.E.; Rajkovic, A.; Matzuk, M.M. TEX14 is essential for intercellular bridges and fertility in male mice. Proc. Natl. Acad. Sci. USA 2006, 103, 4982–4987. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pan, B.; Fei, X.; Hu, Y.; Yang, M.; Yu, H.; Li, J.; Xiong, X. The Biological Characteristics and Differential Expression Patterns of TSSK1B Gene in Yak and Its Infertile Hybrid Offspring. Animals 2023, 13, 320. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Wu, J.; Li, M.; Zuo, F.; Zhang, G. A Comparative Full-Length Transcriptome Analysis Using Oxford Nanopore Technologies (ONT) in Four Tissues of Bovine Origin. Animals 2024, 14, 1646. https://doi.org/10.3390/ani14111646
Liu X, Wu J, Li M, Zuo F, Zhang G. A Comparative Full-Length Transcriptome Analysis Using Oxford Nanopore Technologies (ONT) in Four Tissues of Bovine Origin. Animals. 2024; 14(11):1646. https://doi.org/10.3390/ani14111646
Chicago/Turabian StyleLiu, Xinyue, Jiaxin Wu, Meichen Li, Fuyuan Zuo, and Gongwei Zhang. 2024. "A Comparative Full-Length Transcriptome Analysis Using Oxford Nanopore Technologies (ONT) in Four Tissues of Bovine Origin" Animals 14, no. 11: 1646. https://doi.org/10.3390/ani14111646