
Genome-Wide Gene–Environment Interaction Analysis Identifies Novel Candidate Variants for Growth Traits in Beef Cattle

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Resource and Phenotypes Recording
2.2. Genotyping and Quality Control
2.3. Environmental Factors
2.4. GWEIS
2.5. GWAS
2.6. Gene-Based and Gene-Set Analyses
3. Results
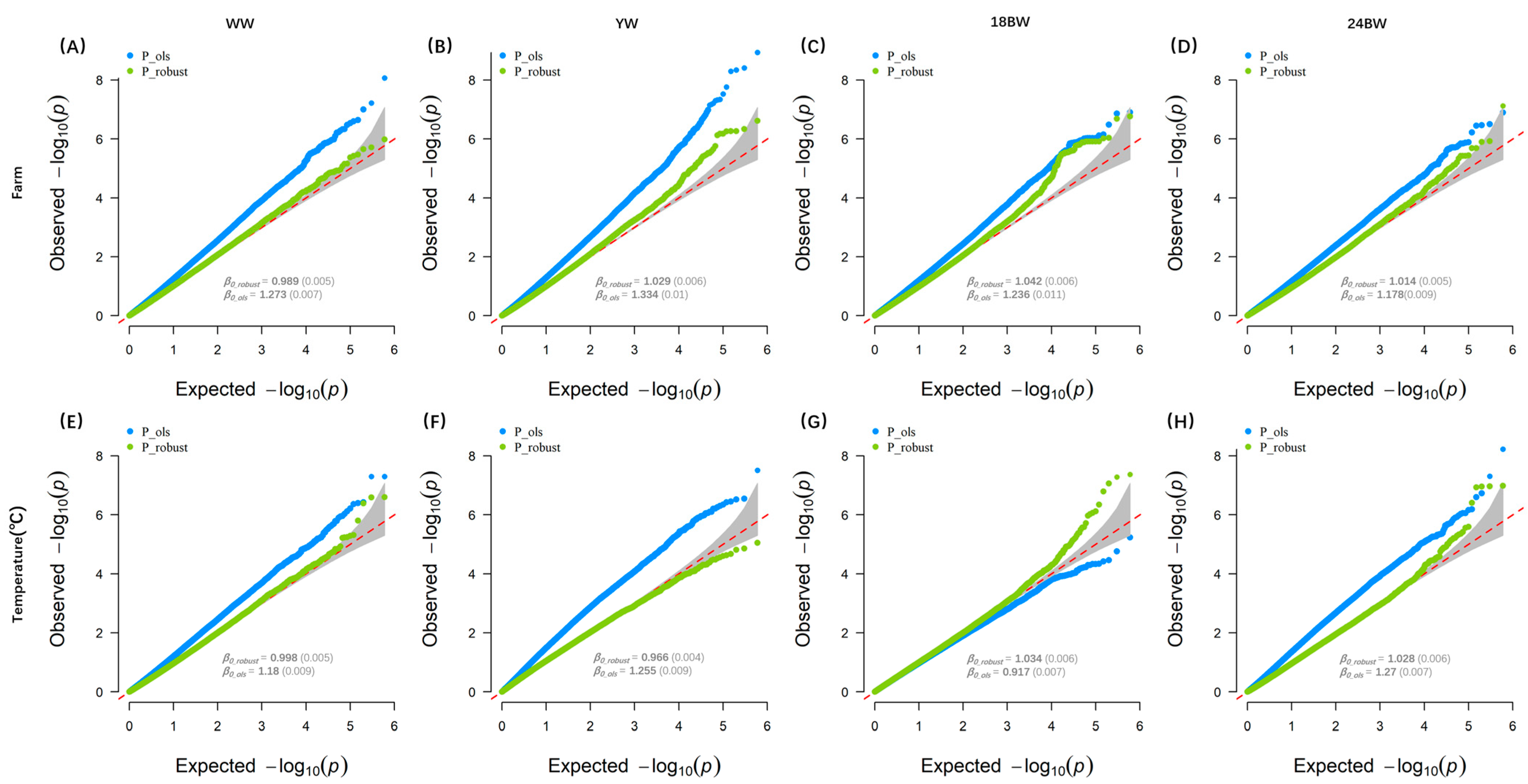
3.1. GWEIS Test Statistics
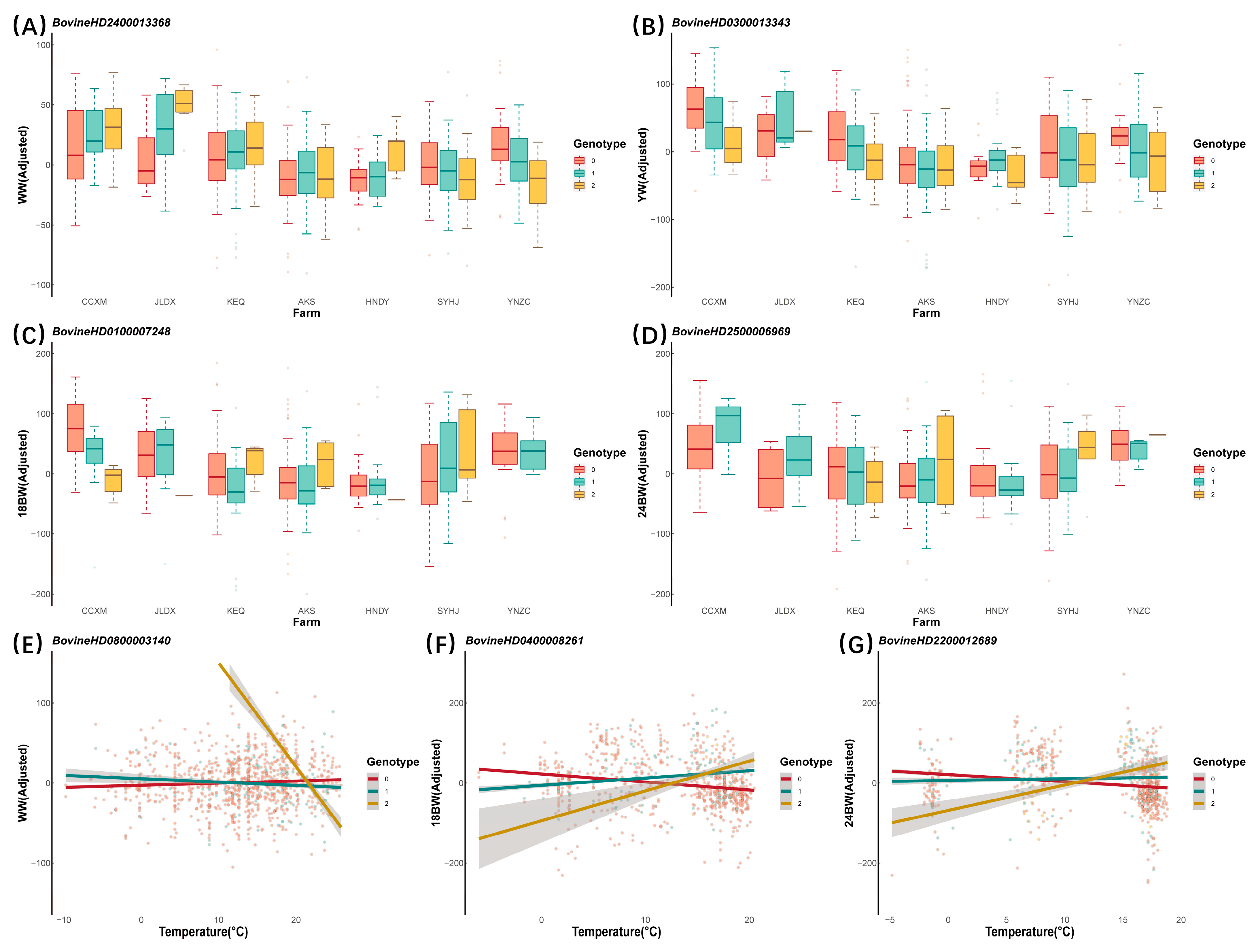
3.2. G × E Interacting SNPs Detected by GWEIS
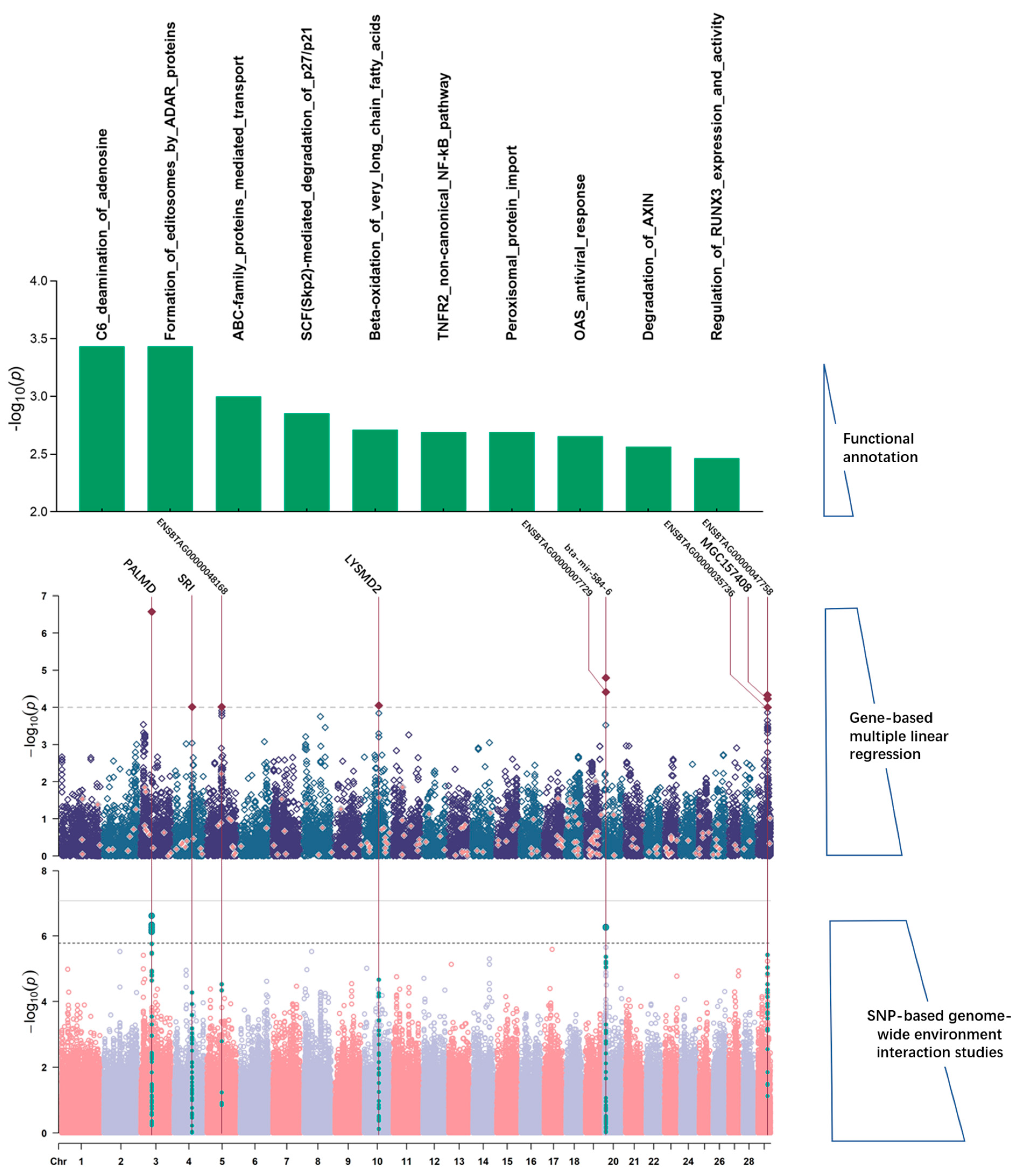
3.3. Genes and Gene Sets Implicated by SNP–Environment Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Utsunomiya, Y.T.; do Carmo, A.S.; Carvalheiro, R.; Neves, H.H.; Matos, M.C.; Zavarez, L.B.; Pérez O’Brien, A.M.; Sölkner, J.; McEwan, J.C.; Cole, J.B.; et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Brito Lopes, F.; da Silva, M.C.; Magnabosco, C.U.; Goncalves Narciso, M.; Sainz, R.D. Selection Indices and Multivariate Analysis Show Similar Results in the Evaluation of Growth and Carcass Traits in Beef Cattle. PLoS ONE 2016, 11, e0147180. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Introduction to quantitative genetics (4th edn). Trends Genet. 1996, 12, 280. [Google Scholar] [CrossRef]
- Seabury, C.M.; Oldeschulte, D.L.; Saatchi, M.; Beever, J.E.; Decker, J.E.; Halley, Y.A.; Bhattarai, E.K.; Molaei, M.; Freetly, H.C.; Hansen, S.L.; et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genom. 2017, 18, 386. [Google Scholar] [CrossRef] [PubMed]
- Bolormaa, S.; Hayes, B.J.; Savin, K.; Hawken, R.; Barendse, W.; Arthur, P.F.; Herd, R.M.; Goddard, M.E. Genome-wide association studies for feedlot and growth traits in cattle. J. Anim. Sci. 2011, 89, 1684–1697. [Google Scholar] [CrossRef] [PubMed]
- Tabery, J.; Griffiths, P.E. Historical and Philosophical Perspectives on Behavioral Genetics and Developmental Science. In Handbook of Developmental Science, Behavior, and Genetics; Hood, K.E., Halpern, C.T., Greenberg, G., Lerner, R.M., Eds.; Blackwell Publishing Ltd.: Chichester, UK, 2010; pp. 39–60. [Google Scholar]
- Caspi, A.; McClay, J.; Moffitt, T.E.; Mill, J.; Martin, J.; Craig, I.W.; Taylor, A.; Poulton, R. Role of genotype in the cycle of violence in maltreated children. Science 2002, 297, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Uher, R.; Zwicker, A. Etiology in psychiatry: Embracing the reality of poly-gene-environmental causation of mental illness. World Psychiatry 2017, 16, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M. Gene-environment interdependence. Dev. Sci. 2007, 10, 12–18. [Google Scholar] [CrossRef]
- Falconer, D.S. Introduction to Quantitative Genetics; Pearson Education India: Delhi, India, 1996. [Google Scholar]
- Williams, J.L.; Bertrand, J.K.; Misztal, I.; Łukaszewicz, M. Genotype by environment interaction for growth due to altitude in United States Angus cattle. J. Anim. Sci. 2012, 90, 2152–2158. [Google Scholar] [CrossRef]
- Bradford, H.L.; Fragomeni, B.O.; Bertrand, J.K.; Lourenco, D.A.L.; Misztal, I. Genetic evaluations for growth heat tolerance in Angus cattle. J. Anim. Sci. 2016, 94, 4143–4150. [Google Scholar] [CrossRef]
- Shi, R.; Brito, L.F.; Liu, A.; Luo, H.; Chen, Z.; Liu, L.; Guo, G.; Mulder, H.; Ducro, B.; van der Linden, A.; et al. Genotype-by-environment interaction in Holstein heifer fertility traits using single-step genomic reaction norm models. BMC Genom. 2021, 22, 193. [Google Scholar] [CrossRef]
- Gillberg, J.; Marttinen, P.; Mamitsuka, H.; Kaski, S. Modelling G×E with historical weather information improves genomic prediction in new environments. Bioinformatics 2019, 35, 4045–4052. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhang, Q.; Ding, X. The superiority of multi-trait models with genotype-by-environment interactions in a limited number of environments for genomic prediction in pigs. J. Anim. Sci. Biotechnol. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Bergen, S.E.; Akula, N.; Song, J.; Hultman, C.M.; Landén, M.; Adli, M.; Alda, M.; Ardau, R.; Arias, B.; et al. Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum. Mol. Genet. 2016, 25, 3383–3394. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K. Factor-analytic models for genotype x environment type problems and structured covariance matrices. Genet. Sel. Evol. 2009, 41, 21. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shi, G.; Nehorai, A. Meta-regression of gene-environment interaction in genome-wide association studies. IEEE Trans. Nanobioscience 2013, 12, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.L.; Holeski, L.M.; Lindroth, R.L. Independent and interactive effects of plant genotype and environment on plant traits and insect herbivore performance: A meta-analysis with Salicaceae. Funct. Ecol. 2019, 33, 422–435. [Google Scholar] [CrossRef]
- Winham, S.J.; Biernacka, J.M. Gene-environment interactions in genome-wide association studies: Current approaches and new directions. J. Child Psychol. Psychiatry Allied Discip. 2013, 54, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Børglum, A.D.; Demontis, D.; Grove, J.; Pallesen, J.; Hollegaard, M.V.; Pedersen, C.B.; Hedemand, A.; Mattheisen, M.; Uitterlinden, A.; Nyegaard, M.; et al. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol. Psychiatry 2014, 19, 325–333. [Google Scholar] [CrossRef]
- Thomas, D. Gene—Environment-wide association studies: Emerging approaches. Nat. Rev. Genet. 2010, 11, 259–272. [Google Scholar] [CrossRef]
- Aguilar, I.; Legarra, A.; Cardoso, F.; Masuda, Y.; Lourenco, D.; Misztal, I. Frequentist p-values for large-scale-single step genome-wide association, with an application to birth weight in American Angus cattle. Genet. Sel. Evol. 2019, 51, 28. [Google Scholar] [CrossRef] [PubMed]
- Kerin, M.; Marchini, J. Inferring Gene-by-Environment Interactions with a Bayesian Whole-Genome Regression Model. Am. J. Hum. Genet. 2020, 107, 698–713. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Madsen, P.; Lund, M.S.; Sorensen, D.; Korsgaard, I.R.; Jensen, J. Bayesian analysis of the linear reaction norm model with unknown covariates. J. Anim. Sci. 2006, 84, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Voorman, A.; Lumley, T.; McKnight, B.; Rice, K. Behavior of QQ-plots and genomic control in studies of gene-environment interaction. PLoS ONE 2011, 6, e19416. [Google Scholar] [CrossRef] [PubMed]
- Werme, J.; van der Sluis, S.; Posthuma, D.; de Leeuw, C.A. Genome-wide gene-environment interactions in neuroticism: An exploratory study across 25 environments. Transl. Psychiatry 2021, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Cheng, S.; Chu, X.; Wen, Y.; Cheng, B.; Liu, L.; Liang, C.; Kafle, O.P.; Jia, Y.; Wu, C.; et al. Associations between electronic devices use and common mental traits: A gene-environment interaction model using the UK Biobank data. Addict. Biol. 2022, 27, e13111. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pan, C.; Cheng, S.; Li, C.; Zhang, H.; Zhang, Z.; Zhang, J.; Yao, Y.; Meng, P.; Yang, X.; et al. The interaction of early life factors and depression-associated loci affecting the age at onset of the depression. Transl. Psychiatry 2022, 12, 294. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.F.; Cai, L. Using heteroskedasticity-consistent standard error estimators in OLS regression: An introduction and software implementation. Behav. Res. Methods 2007, 39, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Almli, L.M.; Duncan, R.; Feng, H.; Ghosh, D.; Binder, E.B.; Bradley, B.; Ressler, K.J.; Conneely, K.N.; Epstein, M.P. Correcting systematic inflation in genetic association tests that consider interaction effects: Application to a genome-wide association study of posttraumatic stress disorder. JAMA Psychiatry 2014, 71, 1392–1399. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Bucan, M. Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 2007, 81, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M.; Schizophrenia Working Group of the Psychiatric Genomics Consortium. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [PubMed]
- White, H. A heteroskedasticity-consistent covariance matrix estimator and a direct test for heteroskedasticity. Econometrica 1980, 48, 817–838. [Google Scholar] [CrossRef]
- Huber, P.J. The behavior of maximum likelihood estimates under nonstandard conditions. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability: Weather Modification, Berkeley, CA, USA, 21 June–18 July 1965; University of California: Berkeley, CA, USA, 1967; p. 221. [Google Scholar]
- Gao, S.K.; Beresford, S.A.; Frank, L.L.; Schreiner, P.J.; Burke, G.L.; Fitzpatrick, A.L. Modifications to the Healthy Eating Index and its ability to predict obesity: The Multi-Ethnic Study of Atherosclerosis. Am. J. Clin. Nutr. 2008, 88, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; van de Vijver, M.J.; Jacquemier, J.; Anderson, T.J.; Osin, P.P.; McGuffog, L.; Easton, D.F. The pathology of familial breast cancer: Predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002, 20, 2310–2318. [Google Scholar] [CrossRef]
- Tchetgen Tchetgen, E.J.; Kraft, P. On the robustness of tests of genetic associations incorporating gene-environment interaction when the environmental exposure is misspecified. Epidemiology 2011, 22, 257–261. [Google Scholar] [CrossRef]
- Liu, D.J.; Leal, S.M. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 2010, 6, e1001156. [Google Scholar] [CrossRef]
- Smith, J.L.; Wilson, M.L.; Nilson, S.M.; Rowan, T.N.; Schnabel, R.D.; Decker, J.E.; Seabury, C.M. Genome-wide association and genotype by environment interactions for growth traits in U.S. Red Angus cattle. BMC Genom. 2022, 23, 517. [Google Scholar]
- Braz, C.U.; Rowan, T.N.; Schnabel, R.D.; Decker, J.E. Genome-wide association analyses identify genotype-by-environment interactions of growth traits in Simmental cattle. Sci. Rep. 2021, 11, 13335. [Google Scholar] [CrossRef]
- Zhang, K.; Rajput, S.K.; Lee, K.B.; Wang, D.; Huang, J.; Folger, J.K.; Knott, J.G.; Zhang, J.; Smith, G.W. Evidence supporting a role for SMAD2/3 in bovine early embryonic development: Potential implications for embryotropic actions of follistatin. Biol. Reprod. 2015, 93, 86. [Google Scholar] [CrossRef]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar] [CrossRef]
- Welle, S.L. Myostatin and muscle fiber size. Focus on “Smad2 and 3 transcription factors control muscle mass in adulthood” and “Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size”. Am. J. Physiol. Cell Physiol. 2009, 296, C1245–C1247. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Coyet, A.; Ollivier, V.; Jeunemaitre, X.; Jondeau, G.; Michel, J.-B.; Vranckx, R. Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms. Cardiovasc. Res. 2011, 89, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Lee, H.-J.; Kim, K.; Caetano-Anolles, K.; Jeong, J.Y.; Park, S.; Oh, Y.K.; Cho, S.; Kim, H. Characterizing milk production related genes in Holstein using RNA-seq. Asian-Australas. J. Anim. Sci. 2016, 29, 343–351. [Google Scholar]
- Martínez-Montes, A.M.; Fernández, A.; Pérez-Montarelo, D.; Alves, E.; Benítez, R.M.; Nuñez, Y.; Óvilo, C.; Ibañez-Escriche, N.; Folch, J.M.; Fernández, A.I. Using RNA-Seq SNP data to reveal potential causal mutations related to pig production traits and RNA editing. Anim. Genet. 2017, 48, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Kalebic, N.; Gilardi, C.; Stepien, B.; Wilsch-Bräuninger, M.; Long, K.R.; Namba, T.; Florio, M.; Langen, B.; Lombardot, B.; Shevchenko, A.; et al. Neocortical Expansion Due to Increased Proliferation of Basal Progenitors Is Linked to Changes in Their Morphology. Cell Stem Cell 2019, 24, 535–550.e9. [Google Scholar] [CrossRef]
- De Weer, A.; Van der Meulen, J.; Rondou, P.; Taghon, T.; Konrad, T.A.; De Preter, K.; Mestdagh, P.; Van Maerken, T.; Van Roy, N.; Jeison, M.; et al. EVI1-mediated down regulation of MIR449A is essential for the survival of EVI1 positive leukaemic cells. Br. J. Haematol. 2011, 154, 337–348. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Xia, J.; Chang, T.; Wang, X.; Miao, J.; Xu, L.; Zhang, L.; Gao, X.; Chen, Y.; Li, J.; et al. Genome-wide association study identifies loci and candidate genes for internal organ weights in Simmental beef cattle. Physiol. Genom. 2018, 50, 523–531. [Google Scholar] [CrossRef]
- Canive, M.; Fernandez-Jimenez, N.; Casais, R.; Vázquez, P.; Lavín, J.L.; Bilbao, J.R.; Blanco-Vázquez, C.; Garrido, J.M.; Juste, R.A.; Alonso-Hearn, M. Identification of loci associated with susceptibility to bovine paratuberculosis and with the dysregulation of the MECOM, eEF1A2, and U1 spliceosomal RNA expression. Sci. Rep. 2021, 11, 313. [Google Scholar] [CrossRef]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.; Dong, Y. Identification and Characterization of circRNA in Longissimus Dorsi of Different Breeds of Cattle. Front. Genet. 2020, 11, 565085. [Google Scholar] [CrossRef]
- Sung, Y.J.; de Las Fuentes, L.; Schwander, K.L.; Simino, J.; Rao, D.C. Gene-smoking interactions identify several novel blood pressure loci in the Framingham Heart Study. Am. J. Hypertens. 2015, 28, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Sevane, N.; Armstrong, E.; Wiener, P.; Pong Wong, R.; Dunner, S.; GemQual Consortium. Polymorphisms in twelve candidate genes are associated with growth, muscle lipid profile and meat quality traits in eleven European cattle breeds. Mol. Biol. Rep. 2014, 41, 4721–4731. [Google Scholar] [CrossRef]
- Martins, R.; Machado, P.C.; Pinto, L.F.B.; Silva, M.R.; Schenkel, F.S.; Brito, L.F.; Pedrosa, V.B. Genome-wide association study and pathway analysis for fat deposition traits in nellore cattle raised in pasture–based systems. J. Anim. Breed. Genet. 2021, 138, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Binder, E.B. Current research trends in early life stress and depression: Review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 2012, 233, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Barr, F.A.; Silljé, H.H.W.; Nigg, E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–440. [Google Scholar] [CrossRef]
- Donahue, H.J.; Li, Z.; Zhou, Z.; Yellowley, C.E. Differentiation of human fetal osteoblastic cells and gap junctional intercellular communication. Am. J. Physiol. Cell Physiol. 2000, 278, C315–C322. [Google Scholar] [CrossRef]
- Cronier, L.; Frendo, J.-L.; Defamie, N.; Pidoux, G.; Bertin, G.; Guibourdenche, J.; Pointis, G.; Malassiné, A. Requirement of gap junctional intercellular communication for human villous trophoblast differentiation. Biol. Reprod. 2003, 69, 1472–1480. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Farms | N | WW | YW | 18 BW | 24 BW | ||||
|---|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | Male | Female | ||
| CCXM | 70 | 258.75 ± 51.38 | 279.31 ± 36.58 | 478.04 ± 48.73 | 391.46 ± 35.55 | 684.5 ± 83.42 | 494.31 ± 47.76 | 839.96 ± 82.45 | 589.08 ± 68.9 |
| JLDX | 35 | 228.74 ± 32.81 | 470.53 ± 61.07 | 637.91 ± 68.14 | 807.55 ± 68.89 | ||||
| KRQ | 238 | 234.54 ± 61.26 | 213.48 ± 30.05 | 444.54 ± 55.11 | 357.13 ± 38.95 | 645.33 ± 94.81 | 459.31 ± 36.73 | 817.68 ± 82.2 | 522 ± 38.38 |
| AKS | 379 | 165.2 ± 64.1 | 152.83 ± 49.64 | 366.12 ± 69.66 | 274.5 ± 57.64 | 594.59 ± 102.3 | 382.82 ± 46.78 | 769.97 ± 155.6 | 447.67 ± 52.59 |
| HNDY | 72 | 184.26 ± 27.37 | 171.93 ± 34.29 | 495.78 ± 43.5 | 475.83 ± 49.14 | 711.5 ± 47.91 | 534.86 ± 47.8 | 880.74 ± 45.06 | 574.5 ± 58.82 |
| SYHJ | 437 | 194.97 ± 24.12 | 208.99 ± 22.86 | 369 ± 39.58 | 285.03 ± 30.32 | 559.67 ± 76.88 | 372.94 ± 35.19 | 740.91 ± 68.86 | 457.19 ± 46.13 |
| YN | 119 | 243.12 ± 56.2 | 232.91 ± 41.74 | 458.17 ± 47.55 | 398.09 ± 30.34 | 685.6 ± 30.9 | 506.43 ± 50.12 | 814.43 ± 54.47 | 588.17 ± 58.02 |
| Total | 1350 | 1187 | 959 | 836 | 777 | ||||
| Environment | Trait | SNP | BTA 1 | POS 2 | Gene | PGWEIS | PGWAS |
|---|---|---|---|---|---|---|---|
| Farms | WW | BovineHD0300020017 | 3 | 67,574,825 | AK5 | 1.03 × 10−6 | 0.614 |
| BovineHD2400013368 | 24 | 47,983,708 | SMAD2 | 1.11 × 10−6 | 0.243 | ||
| YW | BovineHD0300013343 | 3 | 43,785,038 | PALMD | 2.45 × 10−7 | 0.975 | |
| BovineHD2000001962 | 20 | 6,217,212 | MIR584-6 | 5.39 × 10−7 | 0.407 | ||
| 18 BW | BovineHD2400007024 | 24 | 25,954,580 | DSG2, DSG3 | 1.71 × 10−7 | 0.614 | |
| BovineHD0900021567 | 9 | 77,291,417 | NHSL1 | 2.06 × 10−7 | 0.853 | ||
| ARS-BFGL-NGS-58606 | 21 | 34,730,593 | CYP11A1, CCDC33 | 9.11 × 10−7 | 0.271 | ||
| BTB-01643687 | 4 | 8,200,949 | CDK14 | 9.50 × 10−7 | 0.667 | ||
| BovineHD0100028293 | 1 | 99,131,999 | MECOM | 1.23 × 10−6 | 0.011 | ||
| BovineHD1600021531 | 16 | 75,288,520 | - | 1.42 × 10−6 | 0.600 | ||
| BovineHD0100007248 | 1 | 24,519,328 | ROBO2 | 1.59 × 10−6 | 0.249 | ||
| 24 BW | BovineHD2500006969 | 25 | 24,706,966 | - | 7.55 × 10−8 * | 0.982 | |
| BovineHD0200033391 | 2 | 115,898,667 | RHBDD1 | 1.20 × 10−6 | 0.412 | ||
| Temperature | WW | BovineHD0700013465 | 7 | 46,699,643 | - | 2.50 × 10−7 | 0.304 |
| BovineHD0800003140 | 8 | 9,686,754 | HMBOX1 | 4.14 × 10−7 | 0.165 | ||
| BovineHD1900009199 | 19 | 31,126,798 | DNAH9 | 1.57 × 10−6 | 0.277 | ||
| 18 BW | BovineHD0400011664 | 4 | 42,477,921 | - | 4.30 × 10−8 * | 0.368 | |
| BovineHD0800015785 | 8 | 52,604,454 | PCSK5 | 5.20 × 10−8 * | 0.796 | ||
| BovineHD2000001524 | 20 | 4,793,312 | BNIP1 | 8.65 × 10−8 | 0.712 | ||
| BovineHD0600020848 | 6 | 74,944,109 | - | 1.61 × 10−7 | 0.328 | ||
| BovineHD0900023319 | 9 | 83,628,109 | - | 7.59 × 10−7 | 0.857 | ||
| ARS-BFGL-NGS-96591 | 10 | 4,157,289 | - | 8.63 × 10−7 | 0.484 | ||
| BovineHD0800014102 | 8 | 47,106,702 | TRPM3 | 9.57 × 10−7 | 0.761 | ||
| BovineHD0400008261 | 4 | 28,773,445 | - | 1.07 × 10−6 | 0.554 | ||
| 24 BW | BovineHD2200012689 | 22 | 43,774,319 | FLNB | 6.15 × 10−8 * | 0.021 | |
| BovineHD2900006104 | 29 | 21,265,657 | - | 6.42 × 10−8 * | 0.985 | ||
| BovineHD2000005848 | 20 | 19,541,201 | MIR582 | 6.61 × 10−8 * | 0.641 | ||
| BovineHD1900005841 | 19 | 20,432,774 | SARM1, SLC46A1 | 7.05 × 10−8 * | 0.581 | ||
| BovineHD1000001235 | 10 | 4,037,591 | PGGT1B, CCDC112 | 2.46 × 10−7 | 0.729 |
| Environment | Trait | Gene | BTA | POSSTART | POSSTOP | PGWEIS | PGWAS |
|---|---|---|---|---|---|---|---|
| Farms | WW | SMAD2 | 24 | 47,921,047 | 48,072,060 | 4.58 × 10−6 | 0.466 |
| IQCN | 7 | 4,825,678 | 4,954,688 | 1.57 × 10−5 | 0.500 | ||
| CIST1 | 7 | 4,793,442 | 4,960,673 | 5.25 × 10−5 | 0.579 | ||
| YW | PALMD | 3 | 43,638,103 | 43,798,131 | 2.66 × 10−7 * | 0.773 | |
| MIR584-6 | 20 | 6,189,918 | 6,289,993 | 1.59 × 10−5 | 0.231 | ||
| ENSBTAG00000007729 | 20 | 6,155,300 | 6,256,217 | 3.88 × 10−5 | 0.295 | ||
| ENSBTAG00000047758 | 29 | 39,060,430 | 39,165,394 | 4.64 × 10−5 | 0.005 | ||
| MGC157408 | 29 | 39,407,513 | 39,516,695 | 5.89 × 10−5 | 0.024 | ||
| LYSMD2 | 10 | 58,681,531 | 58,794,506 | 8.85 × 10−5 | 0.264 | ||
| ENSBTAG00000048168 | 5 | 59,242,315 | 59,343,254 | 9.68 × 10−5 | 0.302 | ||
| SRI | 4 | 72,724,525 | 72,846,824 | 9.72 × 10−5 | 0.059 | ||
| ENSBTAG00000035736 | 29 | 39,491,478 | 39,600,943 | 9.98 × 10−5 | 0.008 | ||
| 18 BW | MECOM | 1 | 99,040,972 | 99,193,289 | 1.57 × 10−5 | 0.016 | |
| KREMEN2 | 25 | 2,322,471 | 2,427,363 | 3.49 × 10−5 | 0.999 | ||
| 24 BW | ENSBTAG00000046633 | 21 | 69,135,855 | 69,236,382 | 6.31 × 10−5 | 0.669 | |
| Temperature | WW | ABCC10 | 23 | 16,911,898 | 17,031,880 | 2.08 × 10−5 | 0.029 |
| DLK2 | 23 | 16,931,806 | 17,036,060 | 2.10 × 10−5 | 0.029 | ||
| TJAP1 | 23 | 16,951,348 | 17,075,374 | 4.34 × 10−5 | 0.109 | ||
| LRRC73 | 23 | 16,975,858 | 17,078,655 | 6.35 × 10−5 | 0.120 | ||
| YIPF3 | 23 | 16,981,093 | 17,085,360 | 8.49 × 10−5 | 0.127 |
| Environment | Trait | Pathway | PGWEIS | PGWAS |
|---|---|---|---|---|
| Farms | WW | Mitotic Telophase/Cytokinesis | 7.66 × 10−10 * | 0.106 |
| Polo-like kinase-mediated events | 1.27 × 10−5 * | 0.162 | ||
| 24 BW | Mitochondrial Fatty Acid Beta-Oxidation | 5.16 × 10−7 * | 0.288 | |
| Temperature | WW | Reduction in cytosolic Ca++ levels | 2.29 × 10−5 * | 0.906 |
| YW | Formation of annular gap junctions | 9.08 × 10−6 * | 0.230 | |
| Gap junction degradation | 1.87 × 10−5 * | 0.429 | ||
| 24 BW | Keratan sulfate degradation | 1.59 × 10−8 * | 0.069 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, T.; Li, K.; Du, L.; Liang, M.; Qian, L.; Xue, Q.; Qiu, S.; Xu, L.; Zhang, L.; Gao, X.; et al. Genome-Wide Gene–Environment Interaction Analysis Identifies Novel Candidate Variants for Growth Traits in Beef Cattle. Animals 2024, 14, 1695. https://doi.org/10.3390/ani14111695
Deng T, Li K, Du L, Liang M, Qian L, Xue Q, Qiu S, Xu L, Zhang L, Gao X, et al. Genome-Wide Gene–Environment Interaction Analysis Identifies Novel Candidate Variants for Growth Traits in Beef Cattle. Animals. 2024; 14(11):1695. https://doi.org/10.3390/ani14111695
Chicago/Turabian StyleDeng, Tianyu, Keanning Li, Lili Du, Mang Liang, Li Qian, Qingqing Xue, Shiyuan Qiu, Lingyang Xu, Lupei Zhang, Xue Gao, and et al. 2024. "Genome-Wide Gene–Environment Interaction Analysis Identifies Novel Candidate Variants for Growth Traits in Beef Cattle" Animals 14, no. 11: 1695. https://doi.org/10.3390/ani14111695