
Temporal Transcriptome Dynamics of Longissimus dorsi Reveals the Mechanism of the Differences in Muscle Development and IMF Deposition between Fuqing Goats and Nubian Goats
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Management and Sample Collection
2.3. Body Weight, Average Daily Gain, and IMF Collection
2.4. Total RNA Extraction, Quantification, and Qualification
2.5. RNA-Seq Analyses
2.6. Bioinformatic Analyses
2.7. Differential Expression Analyses
2.8. Weighted Gene Coexpression Network Analysis (WGCNA)
2.9. Trend Analysis of DEGs
2.10. Validation by qPCR
2.11. Data Analysis
3. Results
3.1. Production Traits
3.2. Summary of RNA-Seq Results
3.3. Cluster Analysis of 24 LD Muscle Libraries
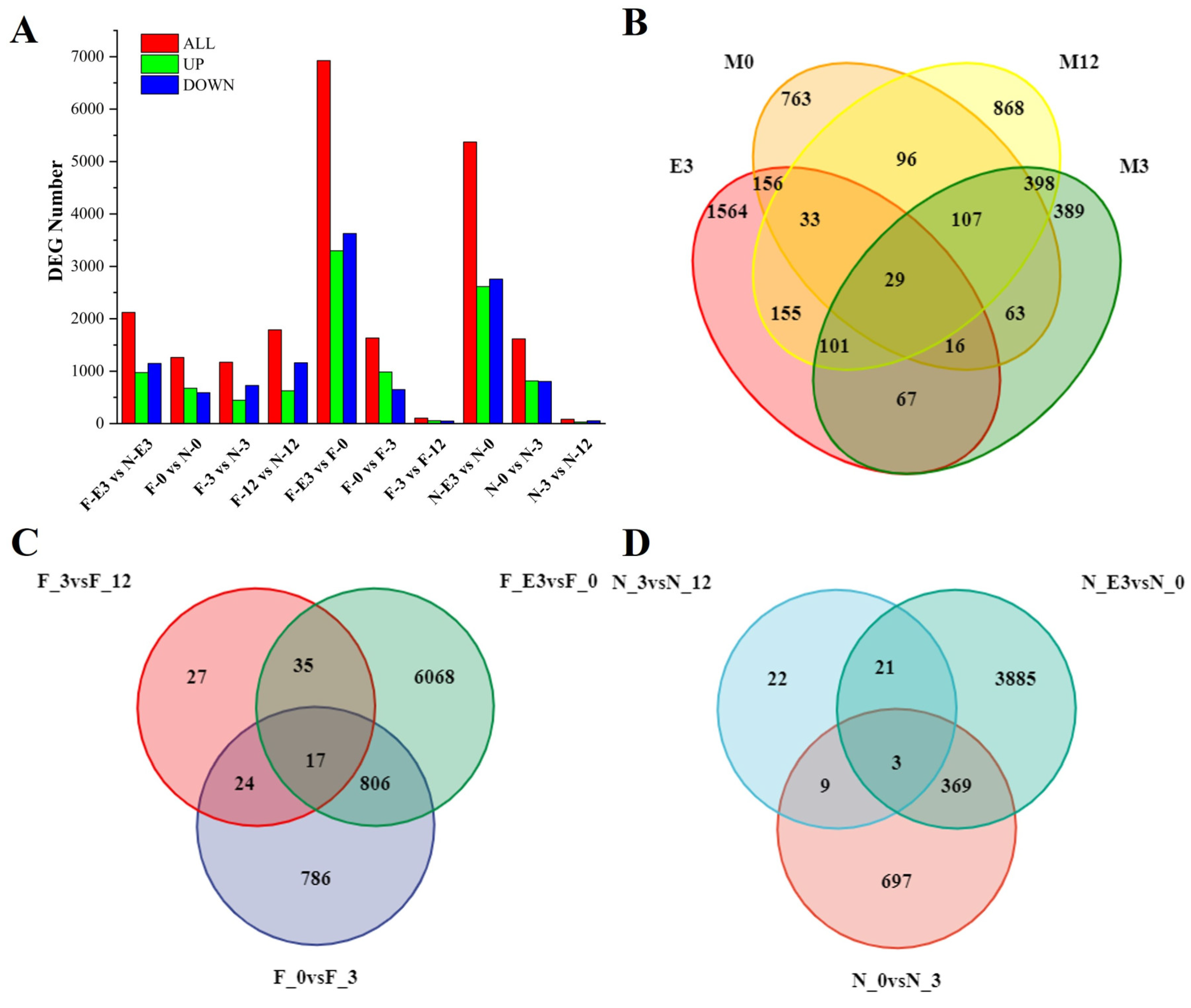
3.4. Identification of DEGs at Different Growth Stages in FQs and NBYs
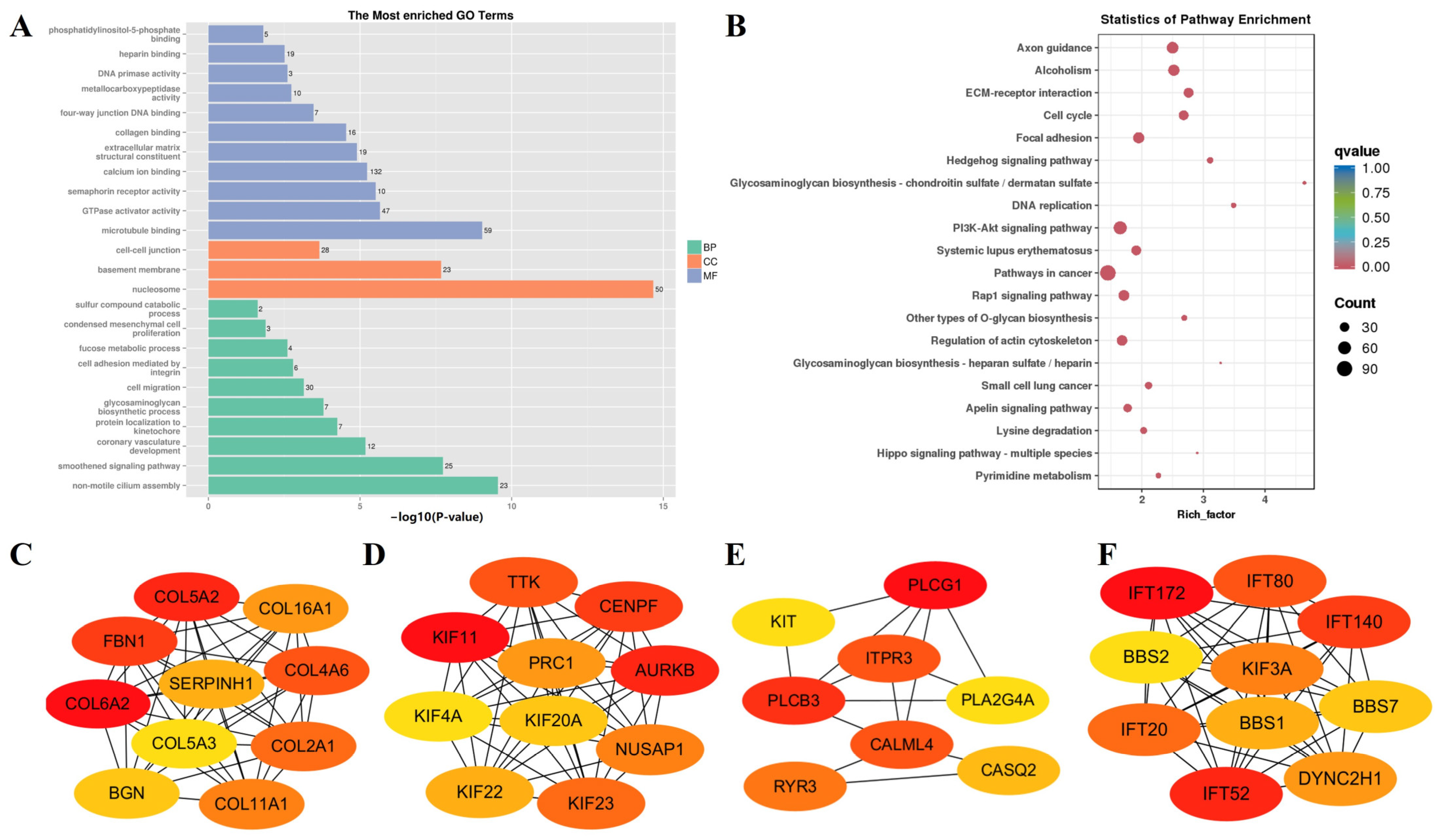
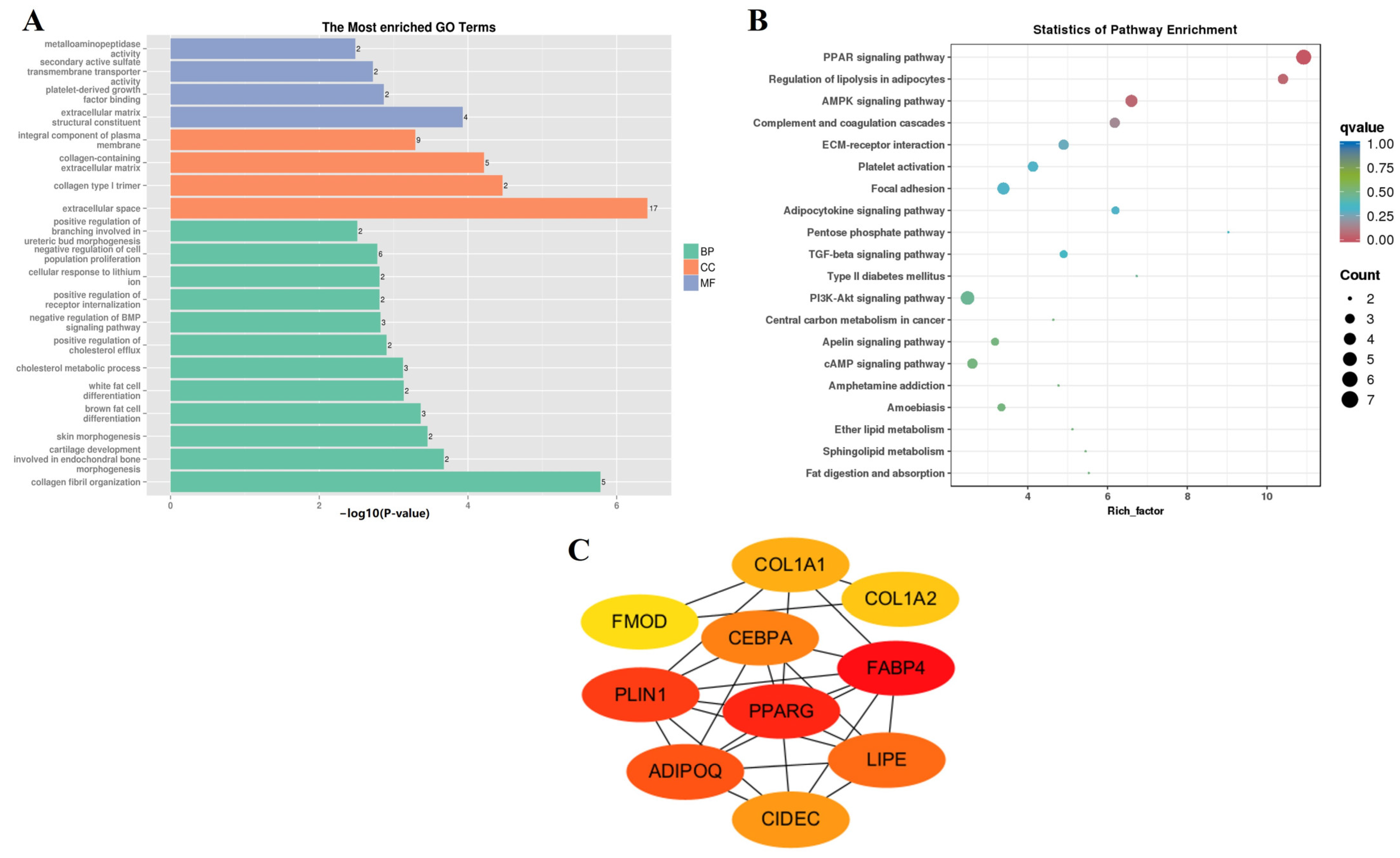
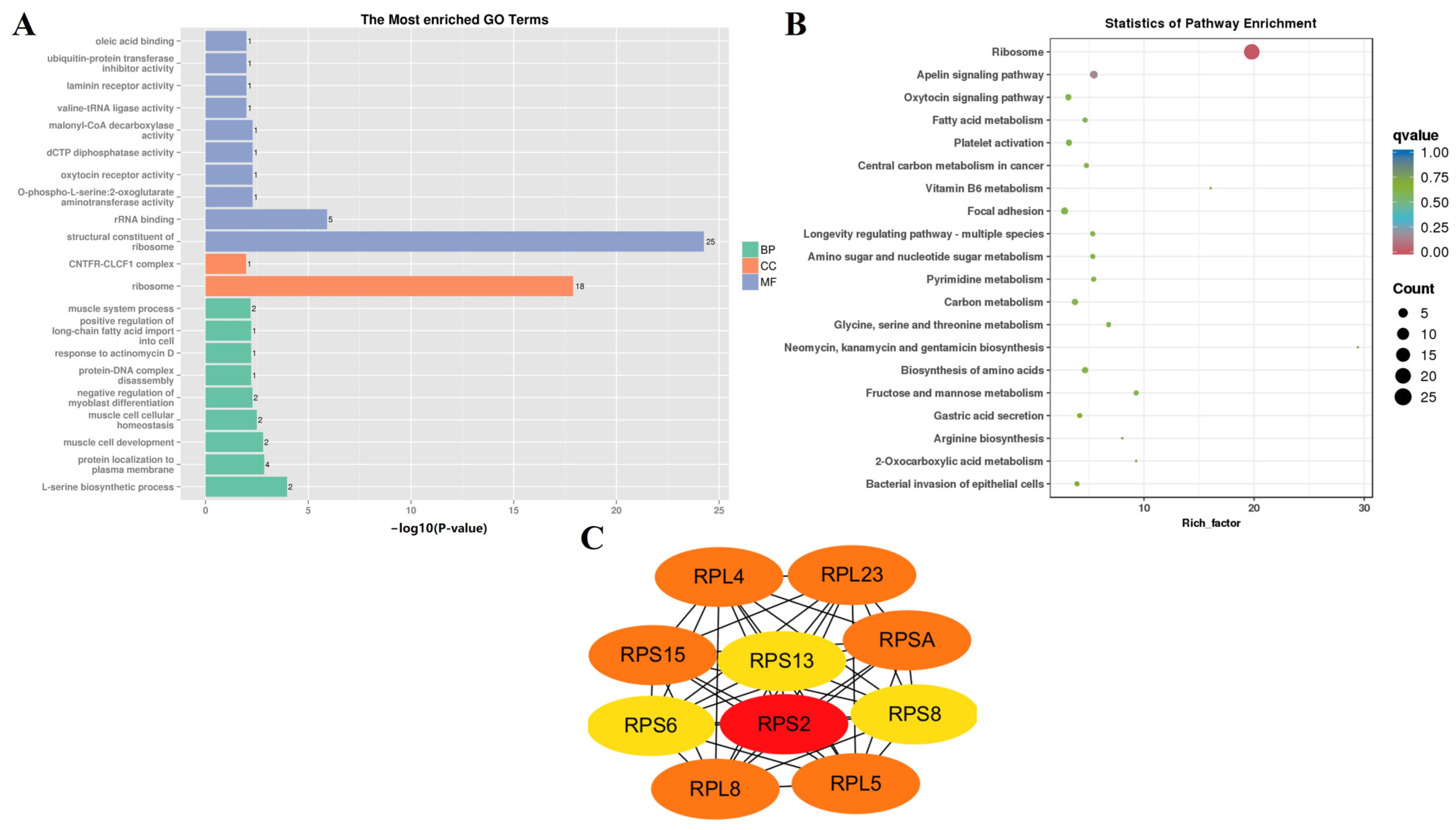
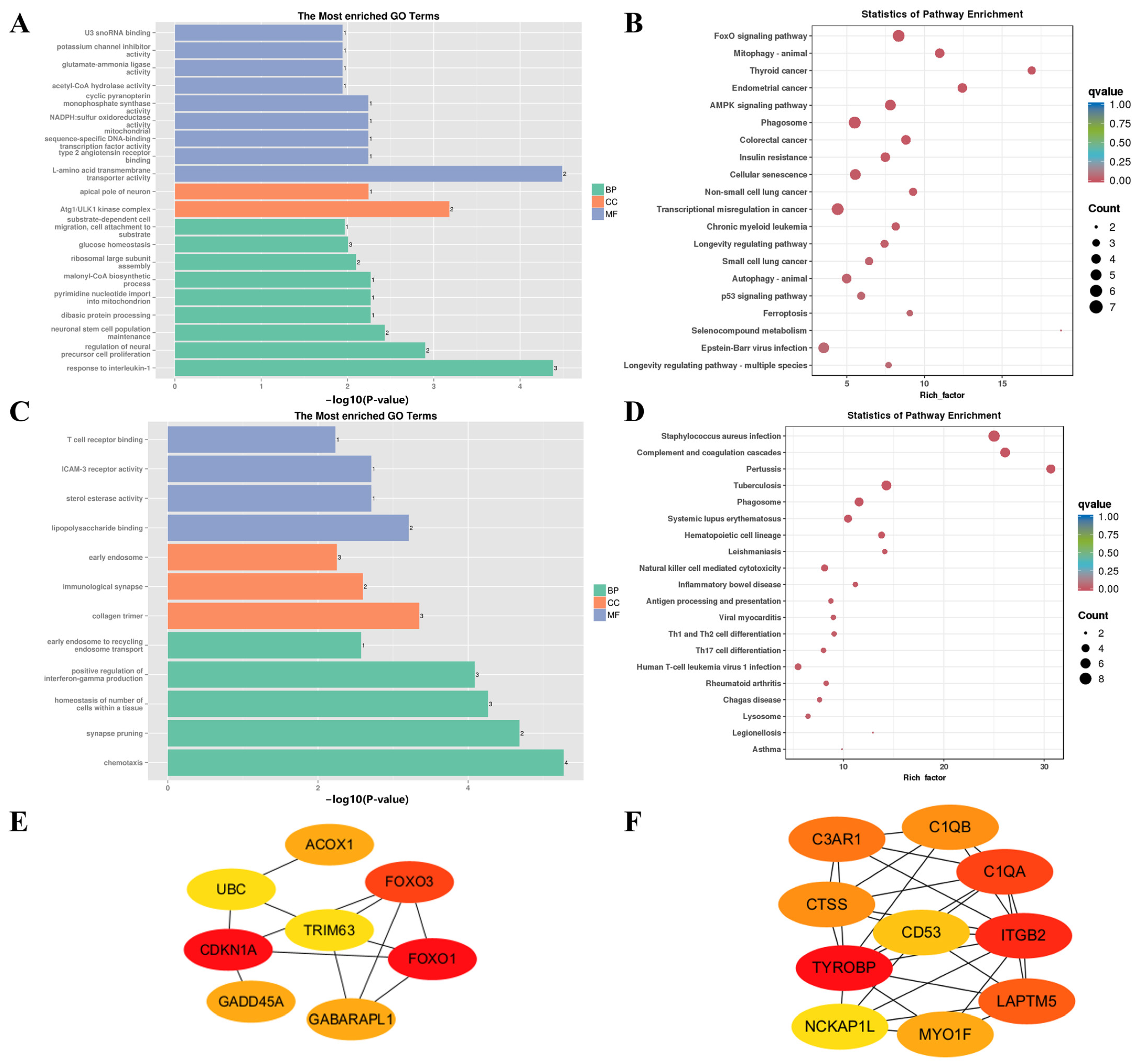
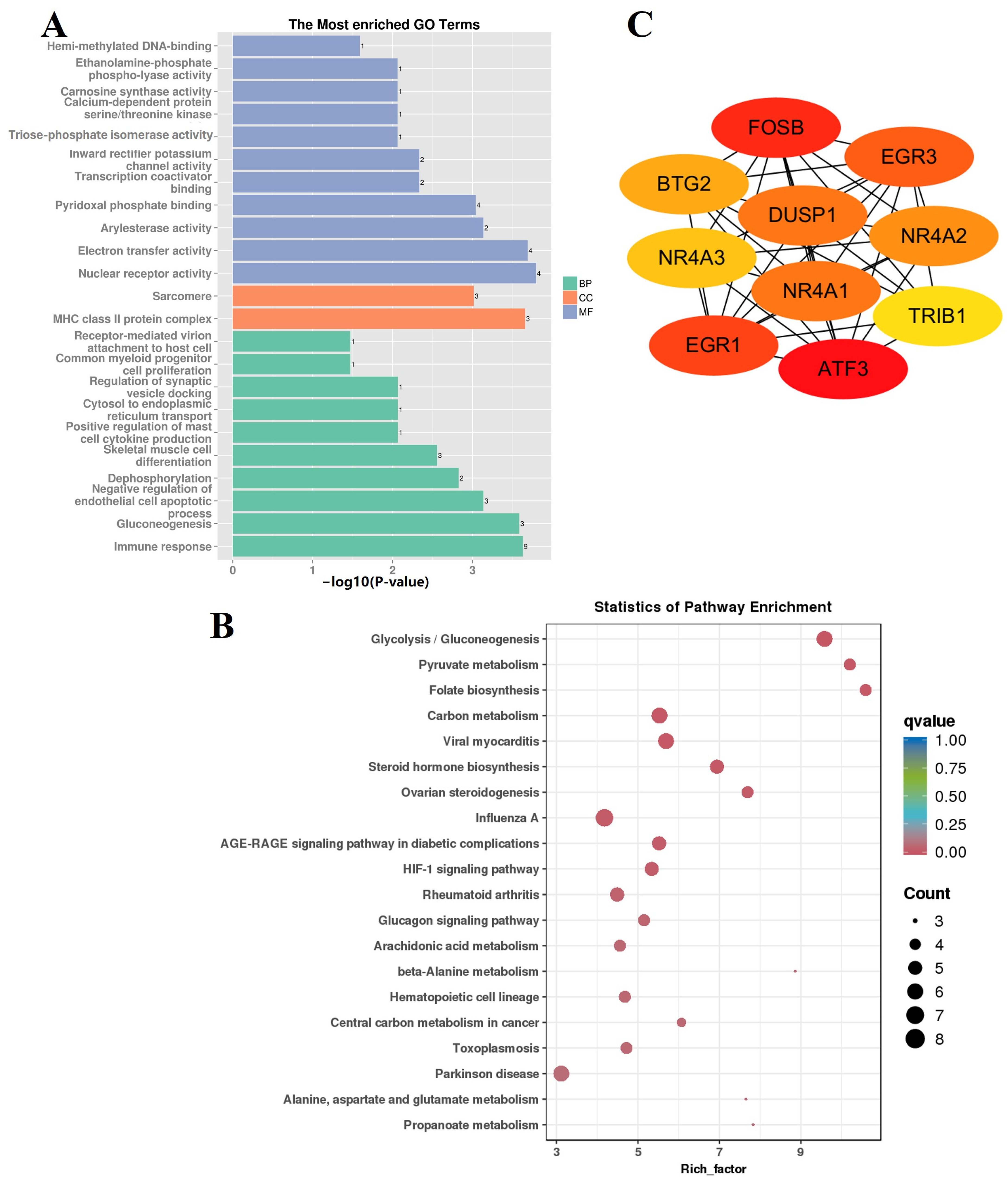
3.5. GO and KEGG Enrichment Analyses of DEGs between FQs and NBYs
3.5.1. GO Enrichment Analyses
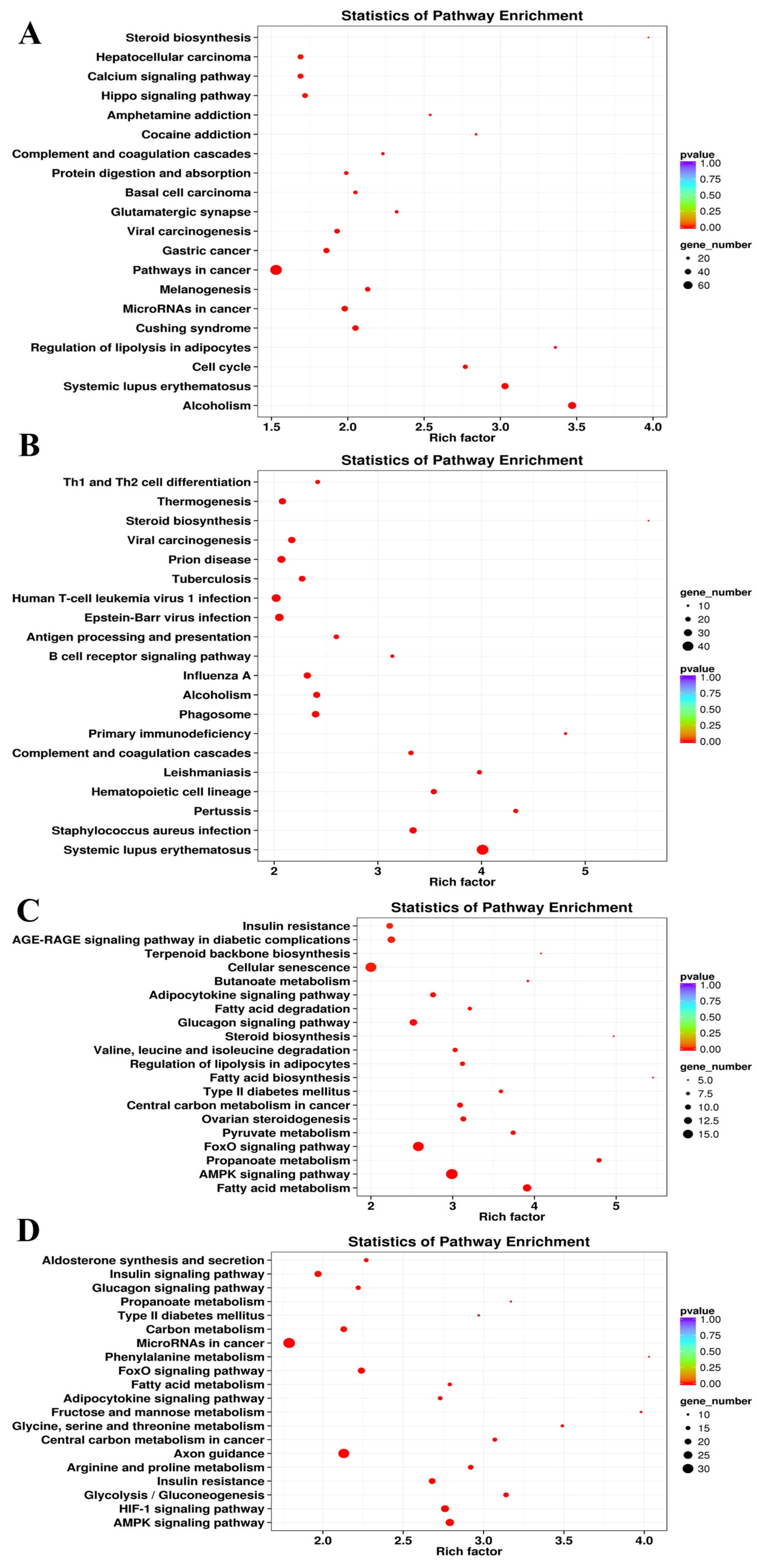
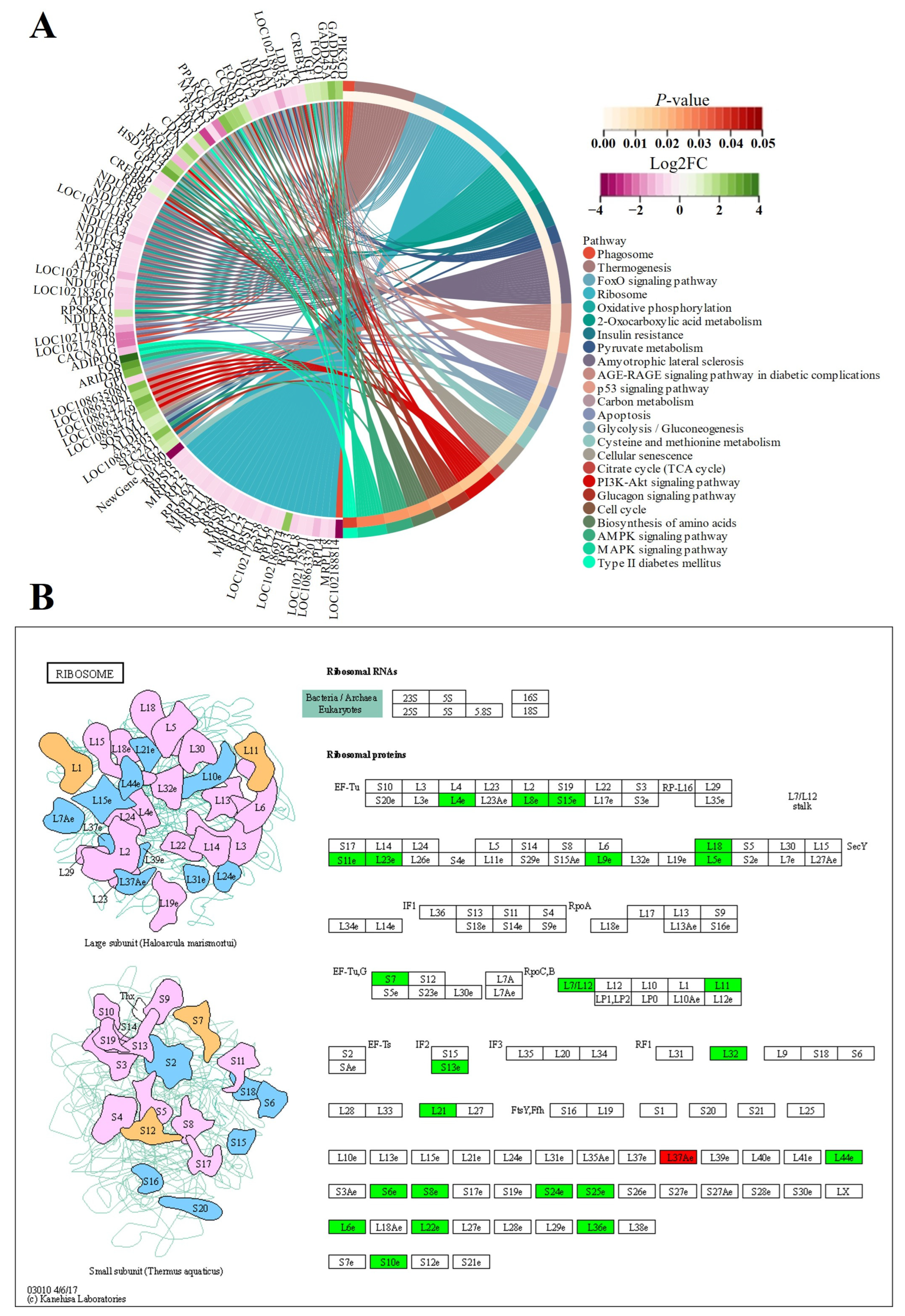
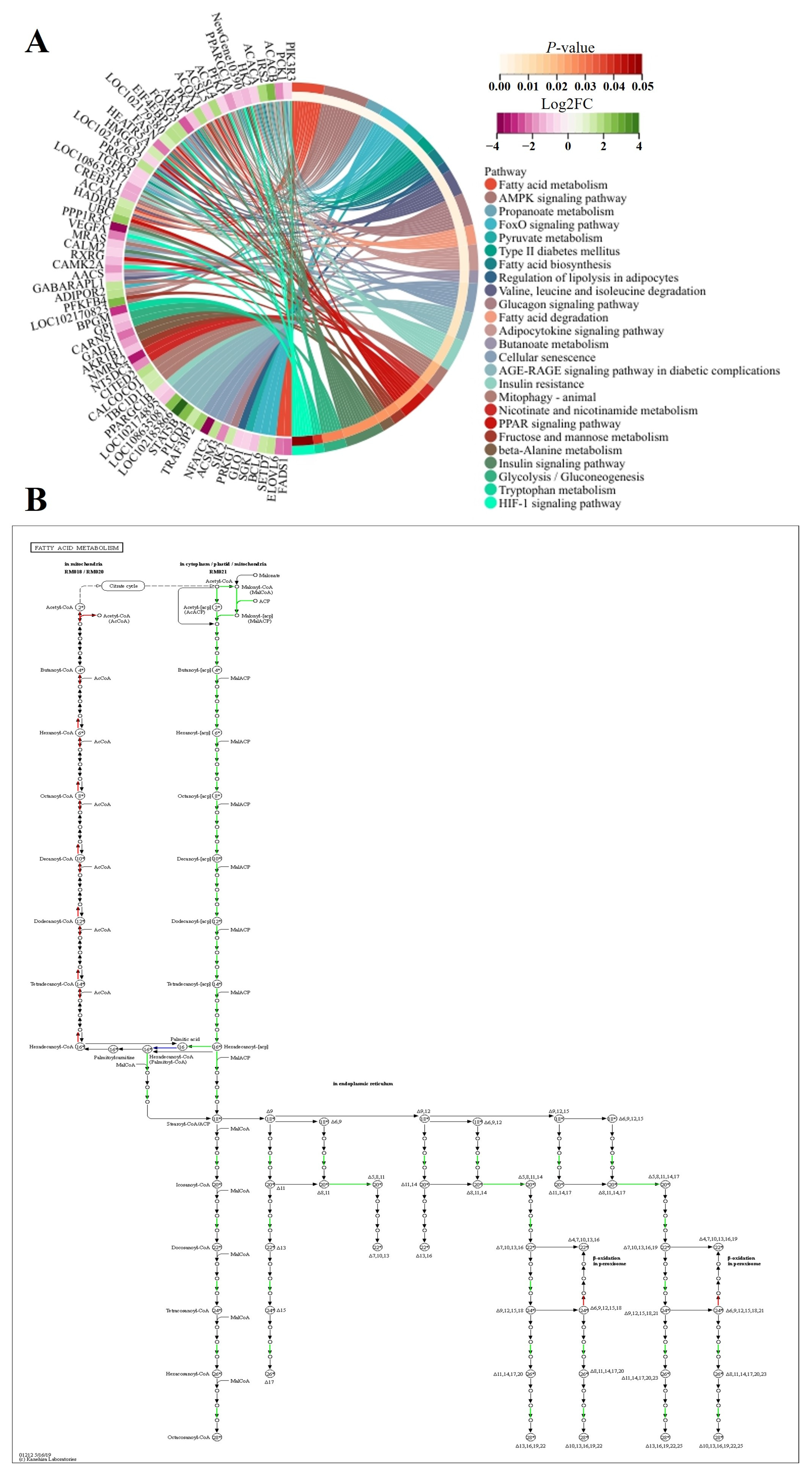
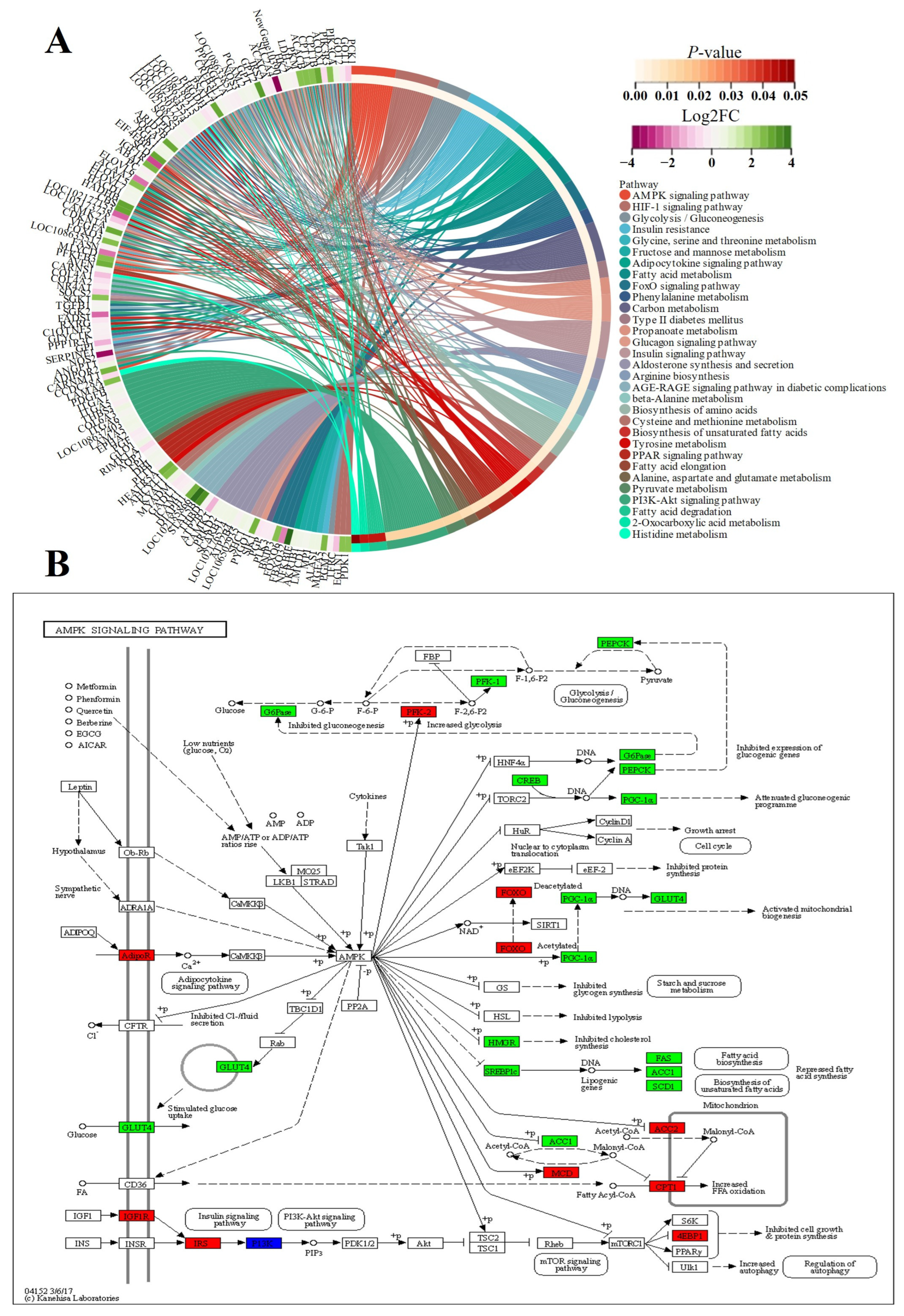
3.5.2. KEGG Enrichment Analyses
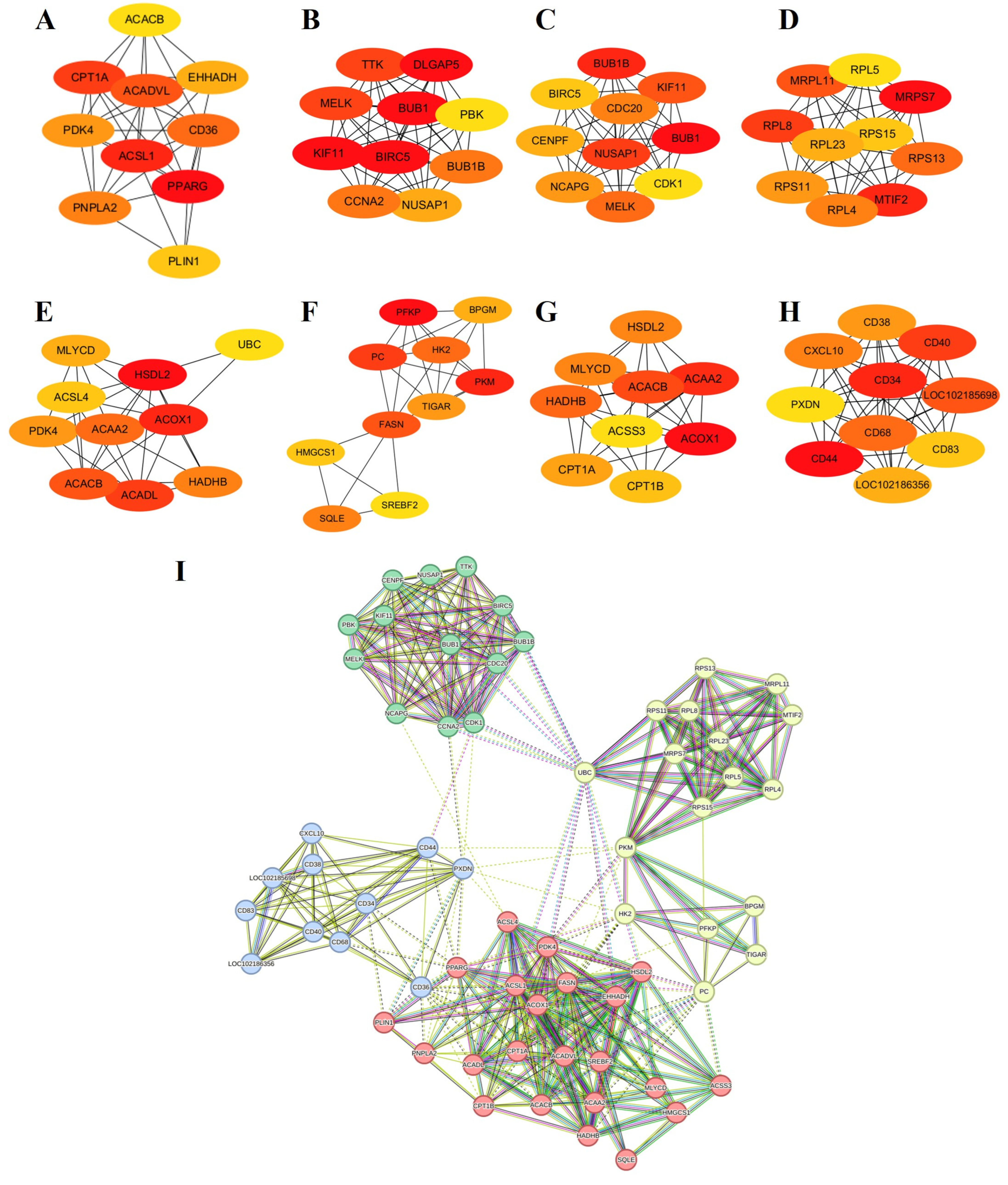
3.5.3. Protein Network Analyses
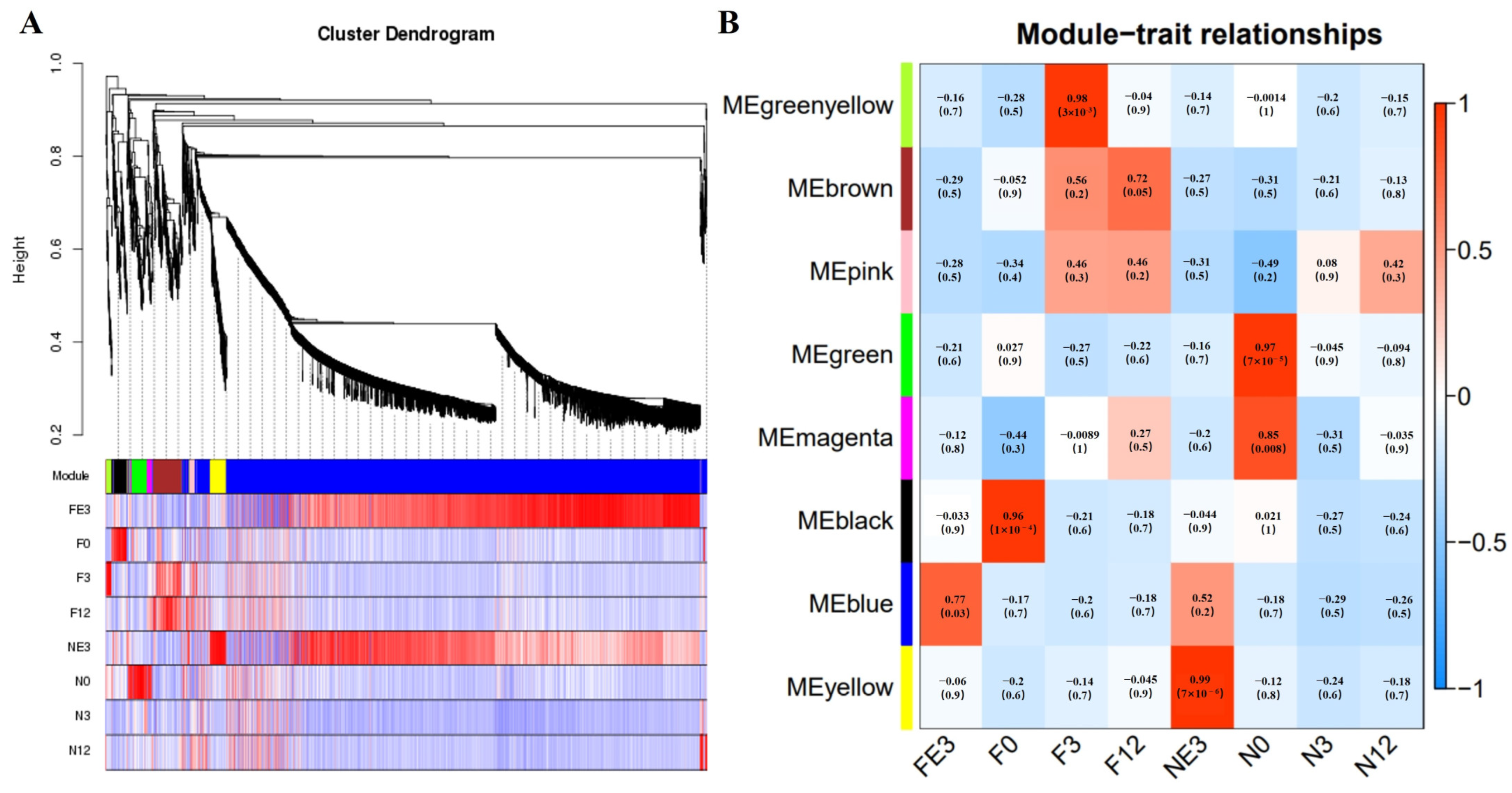
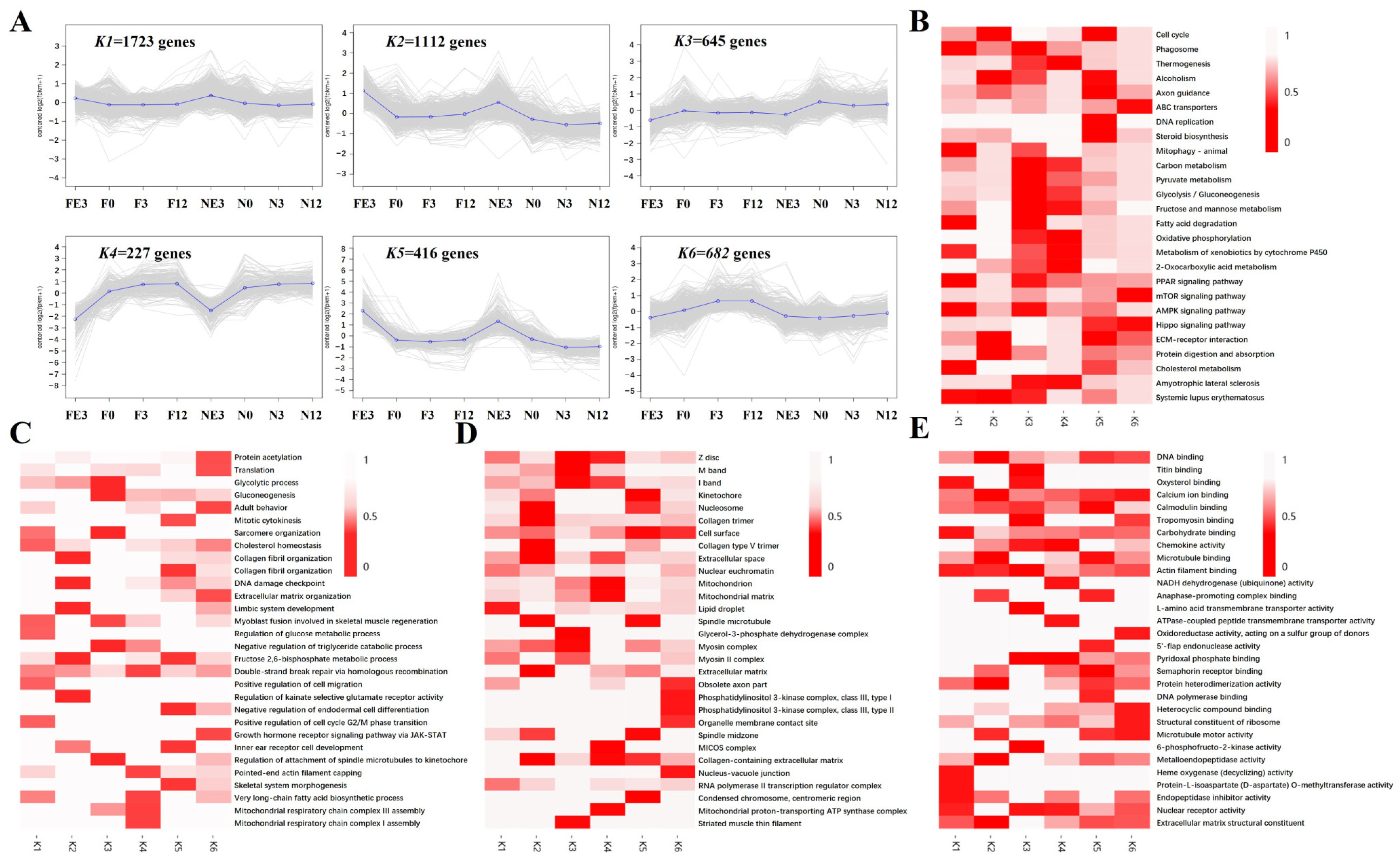
3.6. WGCNA Analyses
3.7. Trend Analysis of DEGs at Four Timepoints between FQs and NBYs
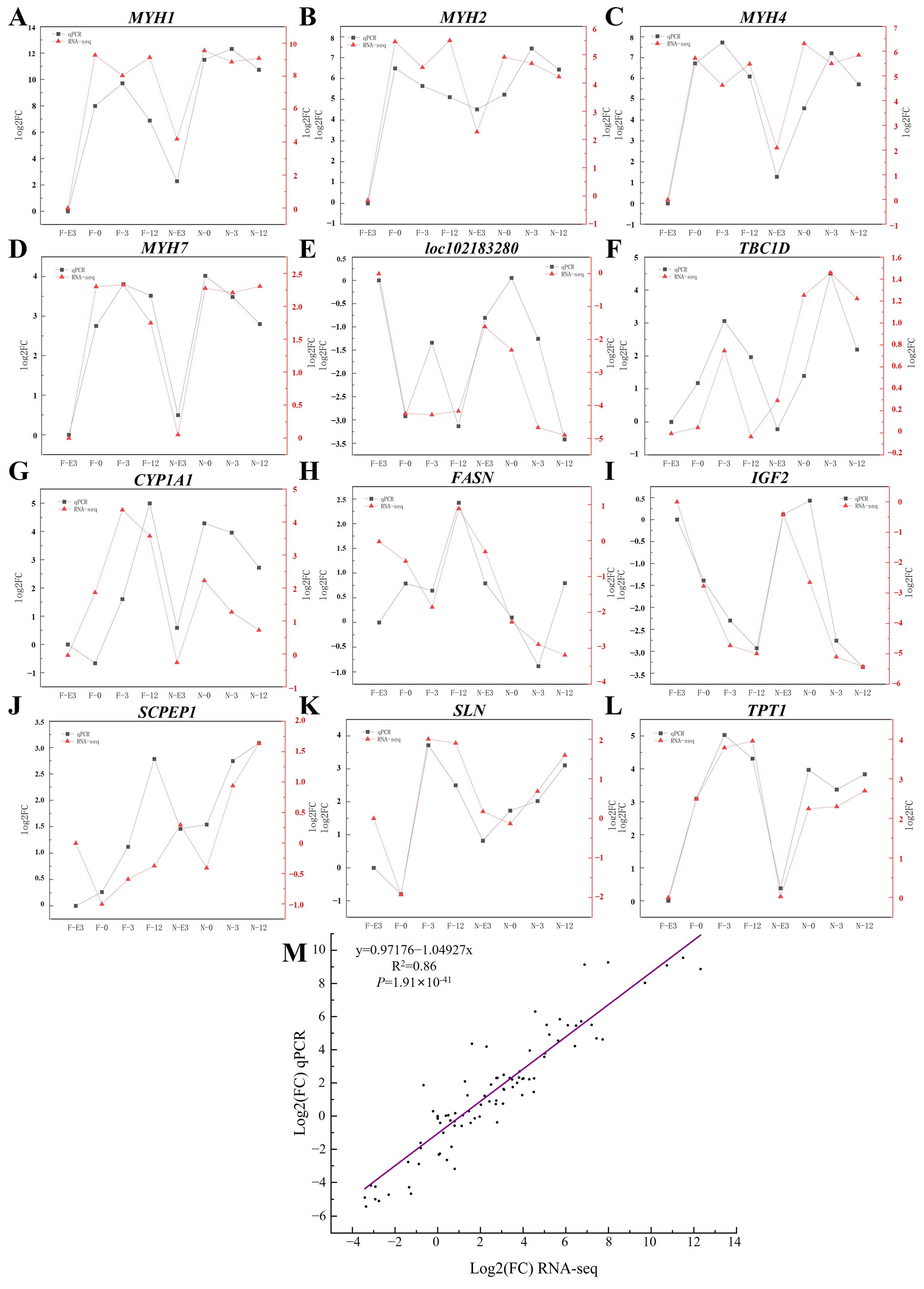
3.8. qPCR Validation
4. Discussion
4.1. Selection of Endogenous Genes for qPCR Verification
4.2. Myogenesis Progresses More Slowly in FQs Than in NBYs
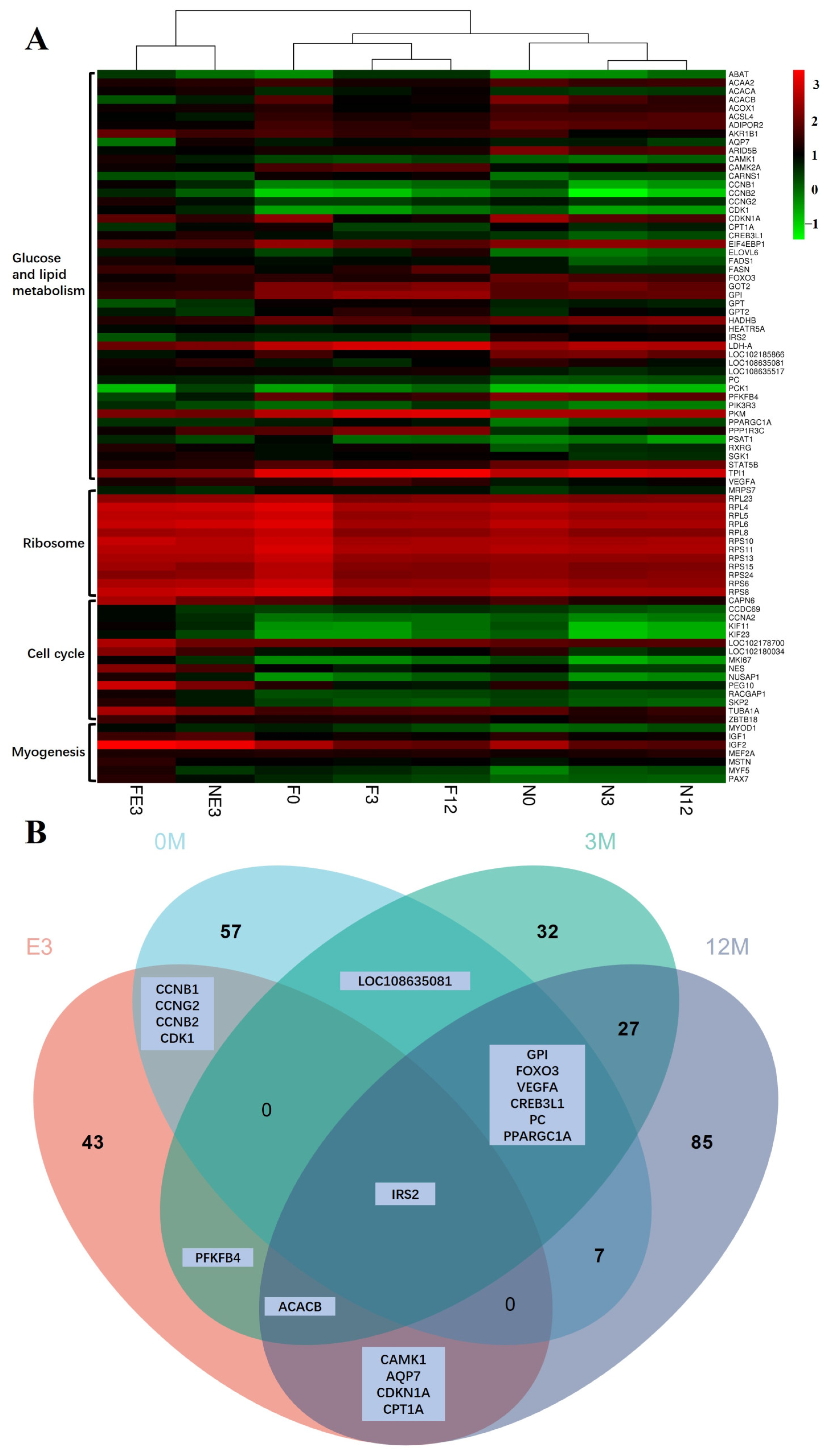
4.3. Insulin Resistance Caused by Low Expression of IRS2 May Determine the Slow Growth Rate of FQs after Weaning
4.4. Differences in Glucose and Lipid Metabolism in LD Determine the Deposition of IMF in FQs and NBYs
4.5. The Unmeasured Differences in Phenotypic Traits of Growth and Meat Quality between FQs and NBYs Limit Our Conclusions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, G.; Zhao, Q.; Lu, J.; Sun, F.; Han, X.; Zhao, J.; Feng, H.; Wang, K.; Liu, C. Insights into the Genetic Diversity of Indigenous Goats and Their Conservation Priorities. Asian-Australas J. Anim. Sci. 2019, 32, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, W.Y.; Wu, X.F.; Huang, Q.L.; Gao, C.F.; Chen, X.Z.; Zhang, X.P. Transcriptome Analysis of Differentially Gene Expression Associated with longissimus doris Tissue in Fuqing Goat and Nubian Black Goat. Sci. Agric. Sin. 2019, 52, 2525–2537. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, L.; Zhao, M.; Zhang, X.; Chen, J.; Zhang, Z.; Cheng, X.; Ren, C. Feeding Regimens Affecting Carcass and Quality Attributes of Sheep and Goat Meat—A Comprehensive Review. Anim. Biosci. 2023, 36, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Liu, Y.; Chen, X.Z.; Zhang, X.P.; Gao, C.F. Growth and Development Indices Measuring and the Growth Curve Fitting Analysis of Fuqing Goats. Fujian J. Agric. Sci. 2015, 30, 545–548. [Google Scholar] [CrossRef]
- Tao, L.; He, X.Y.; Jiang, Y.T.; Lan, R.; Li, M.; Li, Z.M.; Yang, W.F.; Hong, Q.H.; Chu, M.X. Combined Approaches to Reveal Genes Associated with Litter Size in Yunshang Black Goats. Anim. Genet. 2020, 51, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhu, J.; Wang, Y.; Li, Q.; Lin, S. Identification of Differentially Expressed Genes through RNA Sequencing in Goats (Capra hircus) at Different Postnatal Stages. PLoS ONE 2017, 12, e0182602. [Google Scholar] [CrossRef] [PubMed]
- Ncube, K.T.; Dzomba, E.F.; Rosen, B.D.; Schroeder, S.G.; Van Tassell, C.P.; Muchadeyi, F.C. Differential Gene Expression and Identification of Growth-Related Genes in the Pituitary Gland of South African Goats. Front. Genet. 2022, 13, 811193. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, S.; Niu, S.; Bi, X.; Qiao, L.; Yang, K.; Liu, J.; Liu, W. Hybrid Sequencing in Different Types of Goat Skeletal Muscles Reveals Genes Regulating Muscle Development and Meat Quality. Animals 2021, 11, 2906. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.S.; Zhang, X.H.; Gu, L.H.; Zhou, H.L.; Rong, G.; Sun, W.P. Identification and Characterization of Genes Related to the Development of Skeletal Muscle in the Hainan Black Goat. Biosci. Biotechnol. Biochem. 2012, 76, 238–244. [Google Scholar] [CrossRef]
- Brand-Saberi, B. Genetic and Epigenetic Control of Skeletal Muscle Development. Ann. Anat.—Anat. Anz. 2005, 187, 199–207. [Google Scholar] [CrossRef]
- Endo, T. Molecular Mechanisms of Skeletal Muscle Development, Regeneration, and Osteogenic Conversion. Bone 2015, 80, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Ganassi, M.; Badodi, S.; Ortuste Quiroga, H.P.; Zammit, P.S.; Hinits, Y.; Hughes, S.M. Myogenin Promotes Myocyte Fusion to Balance Fibre Number and Size. Nat. Commun. 2018, 9, 4232. [Google Scholar] [CrossRef] [PubMed]
- Murach, K.A.; Fry, C.S.; Kirby, T.J.; Jackson, J.R.; Lee, J.D.; White, S.H.; Dupont-Versteegden, E.E.; McCarthy, J.J.; Peterson, C.A. Starring or Supporting Role? Satellite Cells and Skeletal Muscle Fiber Size Regulation. Physiology 2018, 33, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Zheng, Q.; Jing, J.; Sui, M.; Zhu, L.; Li, Y.; Zhang, Y.; Liu, Y.; Fang, F.; Zhang, X. Switches in Transcriptome Functions during Seven Skeletal Muscle Development Stages from Fetus to Kid in Capra hircus. J. Integr. Agric. 2021, 20, 212–226. [Google Scholar] [CrossRef]
- Abhijith, A.; Warner, R.D.; Dunshea, F.R.; Leury, B.J.; Ha, M.; Chauhan, S.S.; Abhijith, A.; Warner, R.D.; Dunshea, F.R.; Leury, B.J.; et al. A Review of Some Aspects of Goat Meat Quality: Future Research Recommendations. Anim. Prod. Sci. 2023, 63, 1361–1375. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Zhang, C.L.; Plath, M.; Fang, X.T.; Lan, X.Y.; Zhou, Y.; Chen, H. Global Transcriptional Profiling of Longissimus Thoracis Muscle Tissue in Fetal and Juvenile Domestic Goat Using RNA Sequencing. Anim. Genet. 2015, 46, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hao, Z.; Wang, J.; Hu, J.; Liu, X.; Li, S.; Ke, N.; Song, Y.; Lu, Y.; Hu, L.; et al. Comparative Transcriptome Profile Analysis of Longissimus Dorsi Muscle Tissues from Two Goat Breeds with Different Meat Production Performance Using RNA-Seq. Front. Genet. 2021, 11, 619399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, H.; Liu, X.; Yang, Q.; Pan, C.; Lei, C.; Dang, R.; Chen, H.; Lan, X. The Muscle Development Transcriptome Landscape of Ovariectomized Goat. R. Soc. Open Sci. 2017, 4, 171415. [Google Scholar] [CrossRef]
- Zou, J.; Shen, Y.; Zou, J.; Yu, J.; Jiang, Y.; Huang, Y.; Jiang, Q. Transcriptome-Wide Study Revealed That N6-Methyladenosine Participates in Regulation Meat Production in Goats. Foods 2023, 12, 1159. [Google Scholar] [CrossRef]
- NY/T 816-2004; Feeding Standard of Meat-Production Sheep and Goat. China Agriculture Press: Beijing, China, 2004.
- Hopkins, D.L.; Clayton, E.H.; Lamb, T.A.; van de Ven, R.J.; Refshauge, G.; Kerr, M.J.; Bailes, K.; Lewandowski, P.; Ponnampalam, E.N. The Impact of Supplementing Lambs with Algae on Growth, Meat Traits and Oxidative Status. Meat Sci. 2014, 98, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile Hidden Markov Models. Bioinformatics. 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching during Cell Differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated Genome Annotation and Pathway Identification Using the KEGG Orthology (KO) as a Controlled Vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gao, C.; Xu, K.; Jiang, Y.; Niu, J.; Yin, G.; Wang, C. Transcriptome-Based Analysis of Resistance Mechanism to Black Point Caused by Bipolaris sorokiniana in Wheat. Sci. Rep. 2021, 11, 6911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.-D.; Liu, X.; Li, Y.-S.; Ding, J.-P.; Zhang, X.-R.; Zhang, Y.-H. Reference Gene Screening for Analyzing Gene Expression Across Goat Tissue. Asian-Australas J. Anim. Sci. 2013, 26, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.E.; Cherupanakkal, C.; Catherine, M.; Kadhiravan, T.; Parameswaran, N.; Rajendiran, S.; Pillai, A.B. Endogenous Gene Selection for Relative Quantification PCR and IL6 Transcript Levels in the PBMC’s of Severe and Non-Severe Dengue Cases. BMC Res. Notes 2018, 11, 550. [Google Scholar] [CrossRef] [PubMed]
- Najafpanah, M.J.; Sadeghi, M.; Bakhtiarizadeh, M.R. Reference Genes Selection for Quantitative Real-Time PCR Using RankAggreg Method in Different Tissues of Capra Hircus. PLoS ONE 2013, 8, e83041. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Zhang, X.; Li, M.; Luo, N.; Zhao, Y. Screening of Candidate Housekeeping Genes in Uterus Caruncle by RNA-Sequence and qPCR Analyses in Different Stages of Goat (Capra hircus). Animals 2023, 13, 1897. [Google Scholar] [CrossRef]
- Niu, G.; Yang, Y.; Zhang, Y.; Hua, C.; Wang, Z.; Tang, Z.; Li, K. Identifying Suitable Reference Genes for Gene Expression Analysis in Developing Skeletal Muscle in Pigs. PeerJ 2016, 4, e2428. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Li, J.; Liu, H.; Ernst, C.W.; Liu, X.; Liu, G.; Xi, Y.; Lei, M. Evaluation of Housekeeping Genes for Normalizing Real-Time Quantitative PCR Assays in Pig Skeletal Muscle at Multiple Developmental Stages. Gene 2015, 565, 235–241. [Google Scholar] [CrossRef]
- Zhao, B.; Tumaneng, K.; Guan, K.-L. The Hippo Pathway in Organ Size Control, Tissue Regeneration and Stem Cell Self-Renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Chen, C.; Hu, Q.; Fu, Y.; Xu, L.; Wang, C.; Liu, Y. Identification of Robust and Key Differentially Expressed Genes during C2C12 Cell Myogenesis Based on Multiomics Data. Int. J. Mol. Sci. 2022, 23, 6002. [Google Scholar] [CrossRef]
- Zhao, X.; Mo, D.; Li, A.; Gong, W.; Xiao, S.; Zhang, Y.; Qin, L.; Niu, Y.; Guo, Y.; Liu, X.; et al. Comparative Analyses by Sequencing of Transcriptomes during Skeletal Muscle Development between Pig Breeds Differing in Muscle Growth Rate and Fatness. PLoS ONE 2011, 6, e19774. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, J.; Liu, H.; Xi, Y.; Xue, M.; Liu, W.; Zhuang, Z.; Lei, M. Dynamic Transcriptome Profiles of Skeletal Muscle Tissue across 11 Developmental Stages for Both Tongcheng and Yorkshire Pigs. BMC Genom. 2015, 16, 377. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, L.; Zhong, T.; Wang, L.; Guo, J.; Dong, Y.; Feng, J.; Song, T.; Li, L.; Zhang, H. The Differential Proliferation and Differentiation Ability of Skeletal Muscle Satellite Cell in Boer and Nanjiang Brown Goats. Small Rumin. Res. 2018, 169, 99–107. [Google Scholar] [CrossRef]
- Ren, H.; Li, L.; Su, H.; Xu, L.; Wei, C.; Zhang, L.; Li, H.; Liu, W.; Du, L. Histological and Transcriptome-Wide Level Characteristics of Fetal Myofiber Hyperplasia during the Second Half of Gestation in Texel and Ujumqin Sheep. BMC Genom. 2011, 12, 411. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-N.; Liu, C.-L.; Zeng, S.-Q.; Liu, C.-B.; Si, W.-J.; Yuan, Y.; Ren, L.-X.; He, Y.-M.; Zhang, W.-Y.; Zhang, H.-Y.; et al. Identification of Differentially Expressed Long Non-Coding RNAs and Messenger RNAs Involved with Muscle Development in Dazu Black Goats through RNA Sequencing. Anim. Biotechnol. 2023, 34, 1305–1313. [Google Scholar] [CrossRef]
- Wang, L.; Cai, B.; Zhou, S.; Zhu, H.; Qu, L.; Wang, X.; Chen, Y. RNA-Seq Reveals Transcriptome Changes in Goats Following Myostatin Gene Knockout. PLoS ONE 2017, 12, e0187966. [Google Scholar] [CrossRef]
- Zhao, X.; Ye, J.; Lin, X.; Xue, H.; Zou, X.; Liu, G.; Deng, M.; Sun, B.; Guo, Y.; Liu, D.; et al. Identification of Key Functional Genes and LncRNAs Influencing Muscle Growth and Development in Leizhou Black Goats. Genes 2023, 14, 881. [Google Scholar] [CrossRef]
- Buckingham, M.; Vincent, S.D. Distinct and Dynamic Myogenic Populations in the Vertebrate Embryo. Curr. Opin. Genet. Dev. 2009, 19, 444–453. [Google Scholar] [CrossRef]
- Long, Y.C.; Cheng, Z.; Copps, K.D.; White, M.F. Insulin Receptor Substrates Irs1 and Irs2 Coordinate Skeletal Muscle Growth and Metabolism via the Akt and AMPK Pathways. Mol. Cell. Biol. 2011, 31, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Tanokashira, D.; Wang, W.; Maruyama, M.; Kuroiwa, C.; White, M.F.; Taguchi, A. Irs2 Deficiency Alters Hippocampus-Associated Behaviors during Young Adulthood. Biochem. Biophys. Res. Commun. 2021, 559, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hao, Z.; Feng, H.; Li, R.; Zhang, R.; Limesand, S.W.; Zhao, Y.; Chen, X. Insulin Resistance and Dyslipidemia in Low-Birth-Weight Goat Kids. Front. Vet. Sci. 2024, 11, 1370640. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; DelCurto-Wyffels, H.; Thomson, J.; Boles, J. Fat Deposition and Fat Effects on Meat Quality—A Review. Animals 2022, 12, 1550. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Albrecht, E.; Teuscher, F.; Kawabata, K.; Sakashita, K.; Iwamoto, H.; Wegner, J. Differences in Muscle and Fat Accretion in Japanese Black and European Cattle. Meat Sci. 2009, 82, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Naveen Kumar, S.; Sudarshan, S.; Fairoze, M.N.; Kaur, M.; Sharma, A.; Girdhar, Y.; Sreesujatha, R.M.; Devatkal, S.K.; Ahlawat, S.; et al. Transcriptome Profiling of Longissimus Thoracis Muscles Identifies Highly Connected Differentially Expressed Genes in Meat Type Sheep of India. PLoS ONE 2019, 14, e0217461. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, H.; Wang, X.; Zhang, Q.; Guo, T.; Hu, L.; Xu, S. Evaluation of the Longissimus Thoracis et Lumborum Muscle Quality of Chaka and Tibetan Sheep and the Analysis of Possible Mechanisms Regulating Meat Quality. Animals 2023, 13, 2494. [Google Scholar] [CrossRef] [PubMed]
- López, M. Hypothalamic AMPK and Energy Balance. Eur. J. Clin. Investig. 2018, 48, e12996. [Google Scholar] [CrossRef]
- Hishinuma, A.; Majima, M.; Kurabayashi, H. Insulin Resistance in Patients with Stroke Is Related to Visceral Fat Obesity and Adipocytokines. J. Stroke Cerebrovasc. Dis. 2008, 17, 175–180. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Pair | DEGs Number | Number of Significantly Enriched GO | Number of GO Using REViGO | ||||
|---|---|---|---|---|---|---|---|
| BP | CC | MF | BP | CC | MF | ||
| F-E3 vs. N-E3 | 1421 | 244 | 92 | 93 | 145 | 56 | 83 |
| F-0 vs. N-0 | 1156 | 284 | 63 | 176 | 158 | 55 | 152 |
| F-3 vs. N-3 | 1121 | 358 | 45 | 113 | 265 | 38 | 99 |
| F-12 vs. N-12 | 1365 | 314 | 41 | 88 | 194 | 31 | 76 |
| Sample Pair | Class | Annotation | GO ID | p-Values |
|---|---|---|---|---|
| F-E3 vs. N-E3 | BP | regulation of attachment of spindle microtubules to kinetochore | GO:0051988 | 1.55 × 10−5 |
| antibacterial humoral response | GO:0019731 | 1.63 × 10−5 | ||
| mitotic cytokinesis | GO:0000281 | 2.25 × 10−5 | ||
| CENP-A containing nucleosome assembly | GO:0034080 | 5.08 × 10−5 | ||
| myoblast fusion involved in skeletal muscle regeneration | GO:0014905 | 0.00017 | ||
| CC | nucleosome | GO:0000786 | 1.76 × 10−19 | |
| kinetochore | GO:0000776 | 3.96 × 10−5 | ||
| I band | GO:0031674 | 6.42 × 10−5 | ||
| collagen-containing extracellular matrix | GO:0062023 | 0.00014 | ||
| insulin-like growth factor ternary complex | GO:0042567 | 0.00038 | ||
| MF | protein heterodimerization activity | GO:0046982 | 1.38 × 10−16 | |
| anaphase-promoting complex binding | GO:0010997 | 0.00030 | ||
| glutamate-gated calcium ion channel activity | GO:0022849 | 0.00296 | ||
| fructose-2,6-bisphosphate 2-phosphatase activity | GO:0004331 | 0.00477 | ||
| creatine kinase activity | GO:0004111 | 0.02604 | ||
| F-0 vs. N-0 | BP | mitochondrial respiratory chain complex III assembly | GO:0034551 | 8.12 × 10−6 |
| complement activation | GO:0006956 | 4.97 × 10−5 | ||
| mitochondrial transcription | GO:0006390 | 0.00011 | ||
| osteoclast differentiation | GO:0030316 | 0.00024 | ||
| apoptotic cell clearance | GO:0043277 | 0.00432 | ||
| CC | nucleosome | GO:0000786 | 7.46 × 10−9 | |
| external side of plasma membrane | GO:0009897 | 0.00042 | ||
| cell surface | GO:0009986 | 0.00301 | ||
| host cell nucleus | GO:0042025 | 0.00556 | ||
| cell–cell junction | GO:0005911 | 0.00544 | ||
| MF | protein heterodimerization activity | GO:0046982 | 1.76 × 10−6 | |
| DNA-binding transcription activator activity, RNA polymerase II-specific | GO:0001228 | 0.00054 | ||
| oxidoreductase activity, acting on a sulfur group of donors | GO:0016667 | 0.00101 | ||
| L-amino acid transmembrane transporter activity | GO:0015179 | 0.00228 | ||
| NAD(P)+-protein-arginine ADP-ribosyltransferase activity | GO:0003956 | 0.00329 | ||
| F-3 vs. N-3 | BP | negative regulation of chemokine production | GO:0032682 | 8.31 × 10−5 |
| sterol biosynthetic process | GO:0016126 | 3.22 × 10−4 | ||
| B cell apoptotic process | GO:0001783 | 7.78 × 10−4 | ||
| outflow tract morphogenesis | GO:0003151 | 0.00165 | ||
| integrin biosynthetic process | GO:0045112 | 0.00191 | ||
| CC | basement membrane | GO:0005604 | 0.00014 | |
| host cell nucleus | GO:0042025 | 0.00295 | ||
| sarcomere | GO:0030017 | 0.00329 | ||
| membrane raft | GO:0045121 | 0.03073 | ||
| PCSK9-LDLR complex | GO:1990666 | 0.03997 | ||
| MF | sulfonylurea receptor activity | GO:0008281 | 0.00163 | |
| L-amino acid transmembrane transporter activity | GO:0015179 | 0.00163 | ||
| fibronectin binding | GO:0001968 | 0.00328 | ||
| beta-galactoside (CMP) alpha-2,3-sialyltransferase activity | GO:0003836 | 0.00478 | ||
| medium-chain-acyl-CoA dehydrogenase activity | GO:0070991 | 0.00478 | ||
| F-12 vs. N-12 | BP | angiogenesis | GO:0001525 | 2.78 × 10−7 |
| gluconeogenesis | GO:0006094 | 3.53 × 10−6 | ||
| negative regulation of myoblast differentiation | GO:0045662 | 0.00018 | ||
| cellular response to leukemia inhibitory factor | GO:1990830 | 0.00045 | ||
| protein localization to plasma membrane | GO:0072659 | 0.00205 | ||
| CC | basement membrane | GO:0005604 | 5.11 × 10−8 | |
| extracellular space | GO:0005615 | 5.88 × 10−6 | ||
| M band | GO:0031430 | 0.00137 | ||
| adherens junction | GO:0005912 | 0.00855 | ||
| collagen trimer | GO:0005581 | 0.01164 | ||
| MF | actin monomer binding | GO:0003785 | 8.29 × 10−5 | |
| DNA-binding transcription repressor activity, RNA polymerase II-specific | GO:0001227 | 0.00012 | ||
| L-ascorbic acid binding | GO:0031418 | 0.00034 | ||
| metalloendopeptidase activity | GO:0004222 | 0.00076 | ||
| purine-nucleoside phosphorylase activity | GO:0004731 | 0.00425 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wu, X.; Xu, Q.; Lan, X.; Li, W. Temporal Transcriptome Dynamics of Longissimus dorsi Reveals the Mechanism of the Differences in Muscle Development and IMF Deposition between Fuqing Goats and Nubian Goats. Animals 2024, 14, 1770. https://doi.org/10.3390/ani14121770
Liu Y, Wu X, Xu Q, Lan X, Li W. Temporal Transcriptome Dynamics of Longissimus dorsi Reveals the Mechanism of the Differences in Muscle Development and IMF Deposition between Fuqing Goats and Nubian Goats. Animals. 2024; 14(12):1770. https://doi.org/10.3390/ani14121770
Chicago/Turabian StyleLiu, Yuan, Xianfeng Wu, Qian Xu, Xianyong Lan, and Wenyang Li. 2024. "Temporal Transcriptome Dynamics of Longissimus dorsi Reveals the Mechanism of the Differences in Muscle Development and IMF Deposition between Fuqing Goats and Nubian Goats" Animals 14, no. 12: 1770. https://doi.org/10.3390/ani14121770
APA StyleLiu, Y., Wu, X., Xu, Q., Lan, X., & Li, W. (2024). Temporal Transcriptome Dynamics of Longissimus dorsi Reveals the Mechanism of the Differences in Muscle Development and IMF Deposition between Fuqing Goats and Nubian Goats. Animals, 14(12), 1770. https://doi.org/10.3390/ani14121770