
Deciphering the Genetic Variation: A Comparative Analysis of Parental and Attenuated Strains of the QXL87 Vaccine for Infectious Bronchitis

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. Next-Generation Sequencing (NGS)
2.3. Prediction of Protein Post-Translational Modification Sites
2.4. Homology Modeling
2.5. Prediction of Protein-Protein Binding Sites
2.6. Prediction of Potential Virulence Sites
3. Results
3.1. NGS Analysis
3.2. Difference Analysis of PTMs
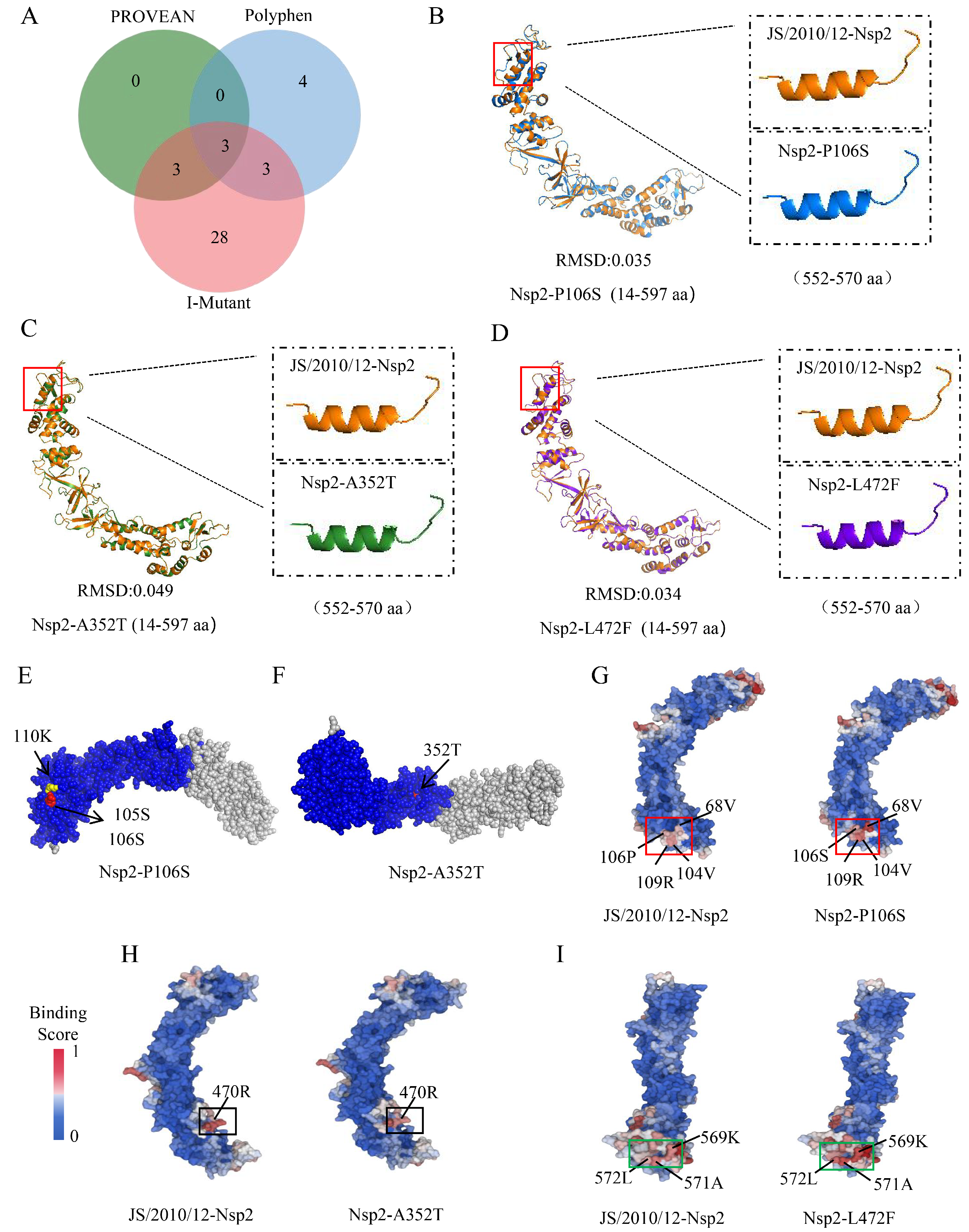
3.3. Difference Analysis of Protein Three-Dimensional Structure
3.4. Difference Analysis of Protein–Protein Binding Sites
3.5. Prediction and Analysis of Virulence Sites on IBV Genome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, J.; Zhao, Y.; Zhang, G. Key Aspects of Coronavirus Avian Infectious Bronchitis Virus. Pathogens 2023, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhao, Y.; Hu, Y.; Zhao, J.; Xue, J.; Zhang, G. The furin-S2’ site in avian coronavirus plays a key role in central nervous system damage progression. J. Virol. 2021, 95, e02447-20. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, Y.; Xu, G.; Zhang, K.; Jia, W.; Sun, Y.; Zhao, J.; Xue, J.; Hu, Y.; Zhang, G. The S2 Subunit of QX-type Infectious Bronchitis Coronavirus Spike Protein Is an Essential Determinant of Neurotropism. Viruses 2019, 11, 972. [Google Scholar] [CrossRef]
- Matthijs, M.G.; Ariaans, M.P.; Dwars, R.M.; van Eck, J.H.; Bouma, A.; Stegeman, A.; Vervelde, L. Course of infection and immune responses in the respiratory tract of IBV infected broilers after superinfection with E. coli. Vet. Immunol. Immunopathol. 2009, 127, 77–84. [Google Scholar] [CrossRef]
- Sid, H.; Benachour, K.; Rautenschlein, S. Co-infection with Multiple Respiratory Pathogens Contributes to Increased Mortality Rates in Algerian Poultry Flocks. Avian Dis. 2015, 59, 440–446. [Google Scholar] [CrossRef]
- Ziebuhr, J. The Coronavirus Replicase. In Coronavirus Replication and Reverse Genetics; Enjuanes, L., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 57–94. [Google Scholar] [CrossRef]
- Deng, X.; van Geelen, A.; Buckley, A.C.; O’Brien, A.; Pillatzki, A.; Lager, K.M.; Faaberg, K.S.; Baker, S.C. Coronavirus Endoribonuclease Activity in Porcine Epidemic Diarrhea Virus Suppresses Type I and Type III Interferon Responses. J. Virol. 2019, 93, e02000-18. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; Hall, D.; Handel, A. Molecular evolution and emergence of avian gammacoronaviruses. Infect. Genet. Evol. 2012, 12, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Ogando, N.S.; Zevenhoven-Dobbe, J.C.; Meer, Y.; Bredenbeek, P.J.; Snijder, E.J. The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS-CoV and SARS-CoV-2. J. Virol. 2020, 94, e01246-20. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Verheije, M.H.; Ulasli, M.; Shaltiël, I.A.; de Vries, L.A.; Reggiori, F.; Rottier, P.J.; de Haan, C.A. Dynamics of coronavirus replication-transcription complexes. J. Virol. 2010, 84, 2134–2149. [Google Scholar] [CrossRef]
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 genome-encoded proteins. Sci. China Life Sci. 2022, 65, 280–294. [Google Scholar] [CrossRef]
- Jordan, B. Vaccination against infectious bronchitis virus: A continuous challenge. Vet. Microbiol. 2017, 206, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Legnardi, M.; Tucciarone, C.M.; Franzo, G.; Cecchinato, M. Infectious Bronchitis Virus Evolution, Diagnosis and Control. Vet. Sci. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Han, Z.; Chen, J.; Liu, X.; Shao, Y.; Kong, X.; Tong, G.; Rong, J. S1 gene sequence heterogeneity of a pathogenic infectious bronchitis virus strain and its embryo-passaged, attenuated derivatives. Avian Pathol. 2007, 36, 231–234. [Google Scholar] [CrossRef]
- Ammayappan, A.; Upadhyay, C.; Gelb, J., Jr.; Vakharia, V.N. Identification of sequence changes responsible for the attenuation of avian infectious bronchitis virus strain Arkansas DPI. Arch. Virol. 2009, 154, 495–499. [Google Scholar] [CrossRef]
- Hodgson, T.; Casais, R.; Dove, B.; Britton, P.; Cavanagh, D. Recombinant infectious bronchitis coronavirus Beaudette with the spike protein gene of the pathogenic M41 strain remains attenuated but induces protective immunity. J. Virol. 2004, 78, 13804–13811. [Google Scholar] [CrossRef] [PubMed]
- Armesto, M.; Cavanagh, D.; Britton, P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS ONE 2009, 4, e7384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, K.; Cheng, J.; Jia, W.; Zhao, Y.; Zhang, G. Replicase 1a gene plays a critical role in pathogenesis of avian coronavirus infectious bronchitis virus. Virology 2020, 550, 1–7. [Google Scholar] [CrossRef]
- van Beurden, S.J.; Berends, A.J.; Krämer-Kühl, A.; Spekreijse, D.; Chenard, G.; Philipp, H.C.; Mundt, E.; Rottier, P.J.M.; Verheije, M.H. Recombinant live attenuated avian coronavirus vaccines with deletions in the accessory genes 3ab and/or 5ab protect against infectious bronchitis in chickens. Vaccine 2018, 36, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cheng, J.; Yan, S.; Jia, W.; Zhang, K.; Zhang, G. S gene and 5a accessory gene are responsible for the attenuation of virulent infectious bronchitis coronavirus. Virology 2019, 533, 12–20. [Google Scholar] [CrossRef]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 gene-based phylogeny of infectious bronchitis virus: An attempt to harmonize virus classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef]
- Yan, K.; Wang, X.; Liu, Z.; Bo, Z.; Zhang, C.; Guo, M.; Zhang, X.; Wu, Y. QX-type infectious bronchitis virus infection in roosters can seriously injure the reproductive system and cause sex hormone secretion disorder. Virulence 2023, 14, 2185380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, K.; Zhang, C.; Guo, M.; Chen, S.; Liao, K.; Bo, Z.; Cao, Y.; Wu, Y. Pathogenicity comparison between QX-type and Mass-type infectious bronchitis virus to different segments of the oviducts in laying phase. Virol. J. 2022, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Tubiana, J.; Schneidman-Duhovny, D.; Wolfson, H.J. ScanNet: An interpretable geometric deep learning model for structure-based protein binding site prediction. Nat. Methods 2022, 19, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Tubiana, J.; Schneidman-Duhovny, D.; Wolfson, H.J. ScanNet: A Web Server for Structure-based Prediction of Protein Binding Sites with Geometric Deep Learning. J. Mol. Biol. 2022, 434, 167758. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R., 3rd; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive characterization of protein-protein interactions perturbed by disease mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef]
- Paniri, A.; Hosseini, M.M.; Akhavan-Niaki, H. First comprehensive computational analysis of functional consequences of TMPRSS2 SNPs in susceptibility to SARS-CoV-2 among different populations. J. Biomol. Struct. Dyn. 2021, 39, 3576–3593. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Zhao, J.; Xie, D.; Huang, X.; Cheng, J.; Guo, Y.; Liu, C.; Ma, Z.; Yang, H.; Zhang, G. Attenuation, safety, and efficacy of a QX-like infectious bronchitis virus serotype vaccine. Vaccine 2018, 36, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Xue, Y.; Wang, J.; Chen, W.; Chen, F.; Bi, Y.; Xie, Q. Development and efficacy of a novel live-attenuated QX-like nephropathogenic infectious bronchitis virus vaccine in China. Vaccine 2015, 33, 1113–1120. [Google Scholar] [CrossRef]
- Listorti, V.; Laconi, A.; Catelli, E.; Cecchinato, M.; Lupini, C.; Naylor, C.J. Identification of IBV QX vaccine markers: Should vaccine acceptance by authorities require similar identifications for all live IBV vaccines? Vaccine 2017, 35, 5531–5534. [Google Scholar] [CrossRef]
- Xu, G.; Ma, S.; Cheng, J.; Zhao, Y.; Zhang, G. An attenuated TW-like infectious bronchitis virus strain has potential to become a candidate vaccine and S gene is responsible for its attenuation. Vet. Microbiol. 2021, 254, 109014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Han, Z.; Zhang, T.; Shao, Y.; Kong, X.; Ma, H.; Liu, S. Genomic characteristics and changes of avian infectious bronchitis virus strain CK/CH/LDL/97I after serial passages in chicken embryos. Intervirology 2014, 57, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Jackwood, M.W.; McKinley, E.T.; Thor, S.W.; Hilt, D.A.; Acevedol, N.D.; Williams, S.M.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.S.; et al. Changes in nonstructural protein 3 are associated with attenuation in avian coronavirus infectious bronchitis virus. Virus Genes 2012, 44, 63–74. [Google Scholar] [CrossRef] [PubMed]
- To, J.; Surya, W.; Fung, T.S.; Li, Y.; Verdià-Bàguena, C.; Queralt-Martin, M.; Aguilella, V.M.; Liu, D.X.; Torres, J. Channel-Inactivating Mutations and Their Revertant Mutants in the Envelope Protein of Infectious Bronchitis Virus. J. Virol. 2017, 91, e02158-16. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liu, Y.; Xia, Y.; Wang, J.; Zheng, X.; Cao, Y. Infectious bronchitis virus nucleocapsid protein suppressed type I interferon production by interfering with the binding of MDA5-dsRNA and interacting with LGP2. Vet. Microbiol. 2023, 284, 109798. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, M.H.; Barta, J.R.; Ojkic, D.; Yoo, D. Complete genomic sequence of turkey coronavirus. Virus Res. 2008, 135, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Liu, M.; Li, W.; Sun, B.; Liu, D.; Wang, G.; Shi, J.; Li, L. Protein post-translational modification in SARS-CoV-2 and host interaction. Front. Immunol. 2022, 13, 1068449. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Thakur, S.S. SARS-CoV-2 Infection Triggers Phosphorylation: Potential Target for Anti-COVID-19 Therapeutics. Front. Immunol. 2022, 13, 829474. [Google Scholar] [CrossRef]
- Davidson, A.D.; Williamson, M.K.; Lewis, S.; Shoemark, D.; Carroll, M.W.; Heesom, K.J.; Zambon, M.; Ellis, J.; Lewis, P.A.; Hiscox, J.A.; et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020, 12, 68. [Google Scholar] [CrossRef]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e1094. [Google Scholar] [CrossRef]
- Gadlage, M.J.; Sparks, J.S.; Beachboard, D.C.; Cox, R.G.; Doyle, J.D.; Stobart, C.C.; Denison, M.R. Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J. Virol. 2010, 84, 280–290. [Google Scholar] [CrossRef]
- de Haan, C.A.; de Wit, M.; Kuo, L.; Montalto-Morrison, C.; Haagmans, B.L.; Weiss, S.R.; Masters, P.S.; Rottier, P.J. The glycosylation status of the murine hepatitis coronavirus M protein affects the interferogenic capacity of the virus in vitro and its ability to replicate in the liver but not the brain. Virology 2003, 312, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Chen, G.; Guo, B.; Cheng, G.; Tang, H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008, 18, 1105–1113. [Google Scholar] [CrossRef]
- Zhu, L.; Fung, S.Y.; Xie, G.; Wong, L.R.; Jin, D.Y.; Cai, Z. Identification of Lysine Acetylation Sites on MERS-CoV Replicase pp1ab. Mol. Cell. Proteom. 2020, 19, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Vann, K.R.; Acharya, A.; Jang, S.M.; Lachance, C.; Zandian, M.; Holt, T.A.; Smith, A.L.; Pandey, K.; Durden, D.L.; El-Gamal, D.; et al. Binding of the SARS-CoV-2 envelope E protein to human BRD4 is essential for infection. Structure 2022, 30, 1224–1232.e1225. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lin, Y.; Zou, C.; Peng, P.; Wu, Y.; Wei, Y.; Liu, Y.; Gong, L.; Cao, Y.; Xue, C. Attenuation and characterization of porcine enteric alphacoronavirus strain GDS04 via serial cell passage. Vet. Microbiol. 2019, 239, 108489. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Moch, C.; Graille, M.; Chapat, C. The SARS-CoV-2 protein NSP2 impairs the silencing capacity of the human 4EHP-GIGYF2 complex. iScience 2022, 25, 104646. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O'Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Fusco, D.N.; Pratt, H.; Kandilas, S.; Cheon, S.S.; Lin, W.; Cronkite, D.A.; Basavappa, M.; Jeffrey, K.L.; Anselmo, A.; Sadreyev, R.; et al. HELZ2 Is an IFN Effector Mediating Suppression of Dengue Virus. Front. Microbiol. 2017, 8, 240. [Google Scholar] [CrossRef]
- Fehr, A.R.; Athmer, J.; Channappanavar, R.; Phillips, J.M.; Meyerholz, D.K.; Perlman, S. The nsp3 macrodomain promotes virulence in mice with coronavirus-induced encephalitis. J. Virol. 2015, 89, 1523–1536. [Google Scholar] [CrossRef]
- Messina, F.; Giombini, E.; Agrati, C.; Vairo, F.; Ascoli Bartoli, T.; Al Moghazi, S.; Piacentini, M.; Locatelli, F.; Kobinger, G.; Maeurer, M.; et al. COVID-19: Viral-host interactome analyzed by network based-approach model to study pathogenesis of SARS-CoV-2 infection. J. Transl. Med. 2020, 18, 233. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhao, P.; Xu, L.D.; Yu, J.Q.; Cai, H.L.; Zhang, C.; Tong, C.; Yang, Y.L.; Xu, P.; Sun, Q.; et al. Enteric coronavirus nsp2 is a virulence determinant that recruits NBR1 for autophagic targeting of TBK1 to diminish the innate immune response. Autophagy 2024, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Netland, J.; DeDiego, M.L.; Zhao, J.; Fett, C.; Álvarez, E.; Nieto-Torres, J.L.; Enjuanes, L.; Perlman, S. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 2010, 399, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Choi, H.J.; Moon, I.S. Mechanistic insights into the deleterious roles of Nasu-Hakola disease associated TREM2 variants. Sci. Rep. 2020, 10, 3663. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Mutation Site | PROVEAN | PolyPhen | I-Mutant (DDG) |
|---|---|---|---|---|
| Nsp2 | A9V | Neutral | Neutral | −0.9 KJ/mol |
| V18I | Neutral | Neutral | −0.39 KJ/mol | |
| P106S | Deleterious | Damaging | −1.42 KJ/mol | |
| H120Q | Neutral | Neutral | −0.37 KJ/mol | |
| V153I | Neutral | Neutral | −0.16 KJ/mol | |
| V155I | Neutral | Neutral | −0.19 KJ/mol | |
| G180S | Neutral | Neutral | −1.33 KJ/mol | |
| T191A | Neutral | Neutral | −1.31 KJ/mol | |
| A197S | Deleterious | Neutral | −0.29 KJ/mol | |
| G231E | Neutral | Damaging | −0.09 KJ/mol | |
| S328P | Neutral | Neutral | −0.91 KJ/mol | |
| A352T | Deleterious | Damaging | −1.24 KJ/mol | |
| A418V | Neutral | Damaging | 0.11 KJ/mol | |
| K448E | Neutral | Neutral | 0.49 KJ/mol | |
| L472F | Deleterious | Damaging | −0.24 KJ/mol | |
| I487V | Neutral | Neutral | −0.95 KJ/mol | |
| A492V | Neutral | Neutral | −0.41 KJ/mol | |
| L560I | Neutral | Neutral | 0.41 KJ/mol | |
| F562S | Neutral | Neutral | −1.35 KJ/mol | |
| L575I | Neutral | Neutral | −0.51 KJ/mol | |
| V590I | Neutral | Neutral | −1.24 KJ/mol | |
| Q591H | Deleterious | Neutral | −0.92 KJ/mol | |
| I601V | Neutral | Neutral | −1.93 KJ/mol | |
| I611T | Neutral | Neutral | −2.39 KJ/mol | |
| V612I | Neutral | Neutral | 0.27 KJ/mol | |
| P616L | Neutral | Neutral | −0.16 KJ/mol | |
| Nsp3 | L16P | Neutral | Neutral | −1.01 KJ/mol |
| D67G | Neutral | Damaging | 0.23 KJ/mol | |
| D115E | Neutral | Neutral | −0.2 KJ/mol | |
| D128E | Neutral | Neutral | 0.76 KJ/mol | |
| Q138E | Neutral | Neutral | 0.55 KJ/mol | |
| D142G | Neutral | Neutral | −0.01 KJ/mol | |
| Q205R | Neutral | Neutral | −0.73 KJ/mol | |
| P235S | Neutral | Neutral | −0.29 KJ/mol | |
| I242V | Neutral | Neutral | −1.18 KJ/mol | |
| T296I | Neutral | Neutral | 0.82KJ/mol | |
| E298K | Neutral | Neutral | −0.22 KJ/mol | |
| E493A | Neutral | Neutral | −0.9 KJ/mol | |
| I554T | Neutral | Neutral | −2.22 KJ/mol | |
| V1577I | Neutral | Neutral | −0.31 KJ/mol | |
| Nsp4 | D490N | Neutral | Damaging | −2.88 KJ/mol |
| S | E64Q | Neutral | Neutral | −0.48 KJ/mol |
| I125T | Neutral | Neutral | −1.08 KJ/mol | |
| S548R | Neutral | Damaging | 0.78 KJ/mol | |
| Y826D | Neutral | Neutral | −1.35 KJ/mol | |
| 3a | Y8F | Neutral | Neutral | 0.78 KJ/mol |
| E | R70I | Neutral | Damaging | −0.52 KJ/mol |
| N | L62F | Neutral | Neutral | 1.11 KJ/mol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Bo, Z.; Zhang, C.; Guo, M.; Wu, Y.; Zhang, X. Deciphering the Genetic Variation: A Comparative Analysis of Parental and Attenuated Strains of the QXL87 Vaccine for Infectious Bronchitis. Animals 2024, 14, 1784. https://doi.org/10.3390/ani14121784
Wang M, Bo Z, Zhang C, Guo M, Wu Y, Zhang X. Deciphering the Genetic Variation: A Comparative Analysis of Parental and Attenuated Strains of the QXL87 Vaccine for Infectious Bronchitis. Animals. 2024; 14(12):1784. https://doi.org/10.3390/ani14121784
Chicago/Turabian StyleWang, Mengmeng, Zongyi Bo, Chengcheng Zhang, Mengjiao Guo, Yantao Wu, and Xiaorong Zhang. 2024. "Deciphering the Genetic Variation: A Comparative Analysis of Parental and Attenuated Strains of the QXL87 Vaccine for Infectious Bronchitis" Animals 14, no. 12: 1784. https://doi.org/10.3390/ani14121784
APA StyleWang, M., Bo, Z., Zhang, C., Guo, M., Wu, Y., & Zhang, X. (2024). Deciphering the Genetic Variation: A Comparative Analysis of Parental and Attenuated Strains of the QXL87 Vaccine for Infectious Bronchitis. Animals, 14(12), 1784. https://doi.org/10.3390/ani14121784