
Transcriptome Analysis Reveals Antioxidant Defense Mechanisms in the Silkworm Bombyx mori after Exposure to Lead
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Silkworm Strain and Lead Exposure
2.2. Determination of Enzyme Activity
2.3. Midgut Collection and RNA Extraction
2.4. Library Construction and High-Throughput Sequencing
2.5. De Novo Assembly and Functional Annotation
2.6. Differential Gene Expression and Gene Enrichment Analysis
2.7. Quantitative Reverse-Transcription PCR (qRT-PCR) Analysis
3. Results and Discussion
3.1. Determination of Enzyme Activity
3.2. De Novo Assembly of the Transcriptome
3.3. Functional Annotation of Unigenes
3.4. Analysis and Identification of Differentially Expressed Genes (DEGs)
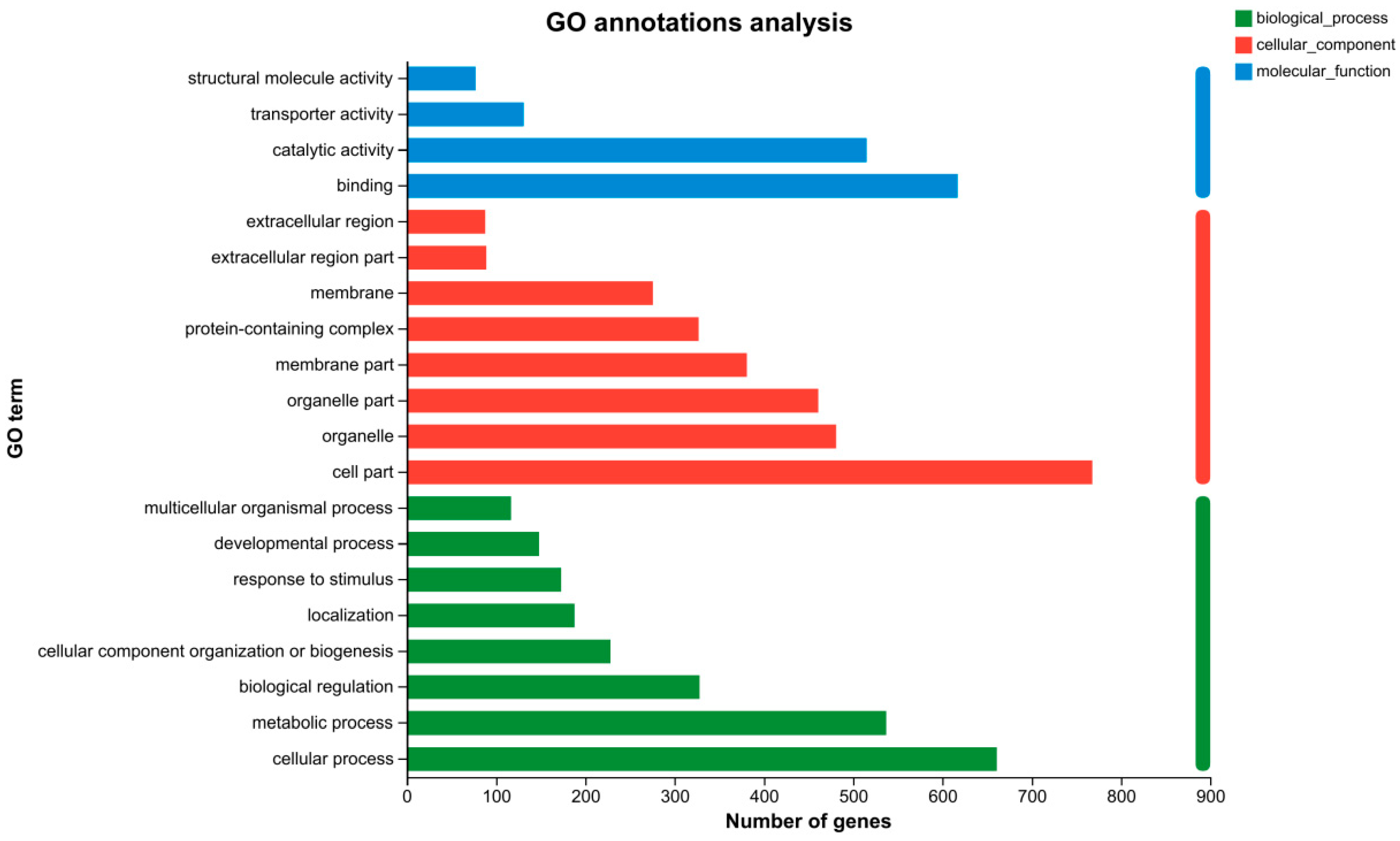
3.5. GO Functional Classification Analysis of the DEGs
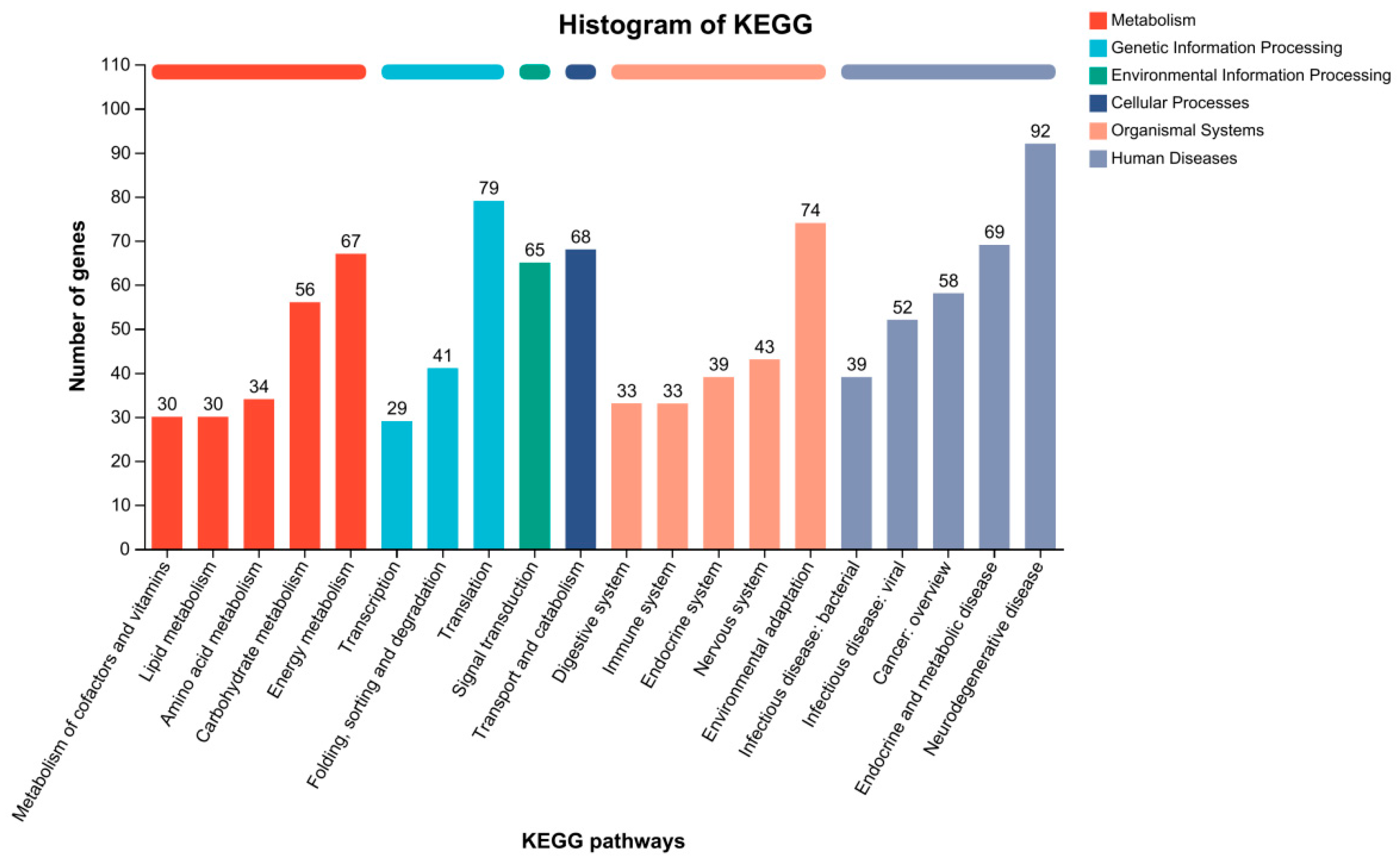
3.6. KEGG Pathway Analysis of DEGs
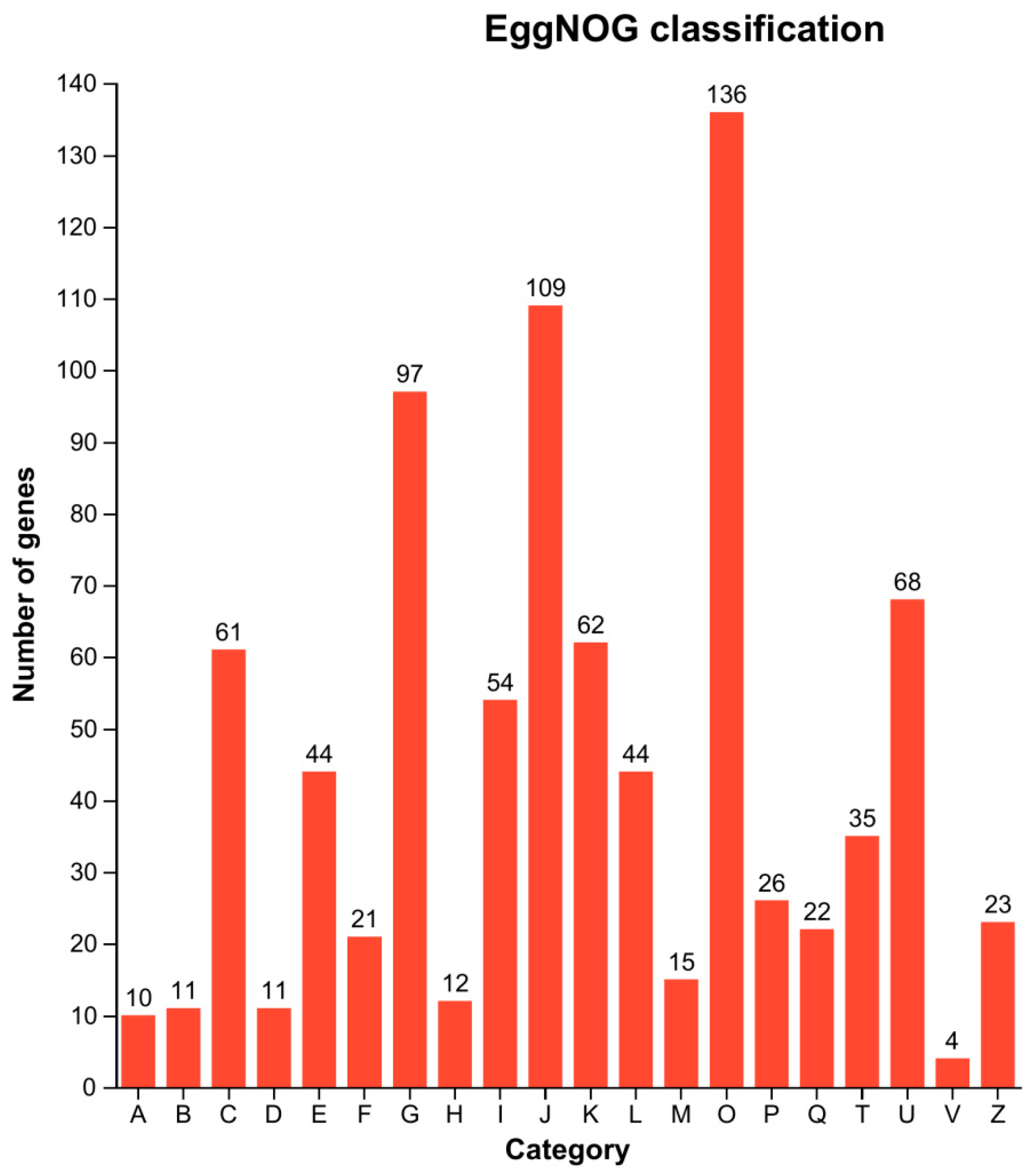
3.7. EggNOG Functional Classification of DEGs
3.8. Validation of Differentially Expressed Genes Using qRT-PCR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodríguez, L.; Ruiz, E.; Alonso-Azcárate, J.; Rincón, J. Heavy metal distribution and chemical speciation in tailings and soils around a Pb-Zn mine in Spain. J. Environ. Manag. 2009, 90, 1106–1116. [Google Scholar] [CrossRef]
- Vareda, J.P.; Valente, A.J.M.; Durães, L. Assessment of heavy metal pollution from anthropogenic activities and remediation strategies: A review. J. Environ. Manag. 2019, 246, 101–118. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metal toxicity and the environment. Exp. Suppl. 2012, 101, 133–164. [Google Scholar] [CrossRef]
- Chen, H.; Teng, Y.; Lu, S.; Wang, Y.; Wang, J. Contamination features and health risk of soil heavy metals in China. Sci. Total Environ. 2015, 512–513, 143–153. [Google Scholar] [CrossRef]
- Tang, W.; Shan, B.; Zhang, H.; Zhang, W.; Zhao, Y.; Ding, Y.; Rong, N.; Zhu, X. Heavy Metal Contamination in the Surface Sediments of Representative Limnetic Ecosystems in Eastern China. Sci. Rep. 2014, 4, 7152. [Google Scholar] [CrossRef]
- Kubota, A. Study of process from Winning Lawsuit to conciliation in disaster area of itai-itai disease—How should we recover from the disaster of land contamination by environmental disruption. Jpn. Archit. Rev. 2020, 3, 231–240. [Google Scholar] [CrossRef]
- Harada, M. Minamata disease: Methylmercury poisoning in Japan caused by environmental pollution. Crit. Rev. Toxicol. 1995, 25, 1–24. [Google Scholar] [CrossRef]
- Whitehead, L.S.; Buchanan, S.D. Childhood Lead Poisoning: A Perpetual Environmental Justice Issue. J. Public Health Manag. Pract. 2019, 25, S115–S120. [Google Scholar] [CrossRef]
- Longman, J.; Ersek, V.; Veres, D. High variability between regional histories of long-term atmospheric Pb pollution. Sci. Rep. 2020, 10, 20890. [Google Scholar] [CrossRef]
- Delile, H.; Blichert-Toft, J.; Goiran, J.P.; Keay, S.; Albarède, F. Lead in ancient Rome’s city waters. Proc. Natl. Acad. Sci. USA 2014, 111, 6594–6599. [Google Scholar] [CrossRef]
- Longman, J.; Veres, D.; Finsinger, W.; Ersek, V. Exceptionally high levels of lead pollution in the Balkans from the Early Bronze Age to the Industrial Revolution. Proc. Natl. Acad. Sci. USA 2018, 115, E5661–E5668. [Google Scholar] [CrossRef]
- Darlington, P.J. Drifting continents and late paleozoic geography. Proc. Natl. Acad. Sci. USA 1964, 52, 1084–1091. [Google Scholar] [CrossRef]
- Weiss, D.; Shotyk, W.; Appleby, P.G.; Kramers, J.D.; Cheburkin, A.K. Atmospheric Pb Deposition since the Industrial Revolution Recorded by Five Swiss Peat Profiles: Enrichment Factors, Fluxes, Isotopic Composition, and Sources. Environ. Sci. Technol. 1999, 33, 1340–1352. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, A.; Cabral-Pinto, M.M.S.; Chaturvedi, A.K.; Shabnam, A.A.; Subrahmanyam, G.; Mondal, R.; Gupta, D.K.; Malyan, S.K.S.; Kumar, S.A.; et al. Lead Toxicity: Health Hazards, Influence on Food Chain, and Sustainable Remediation Approaches. Int. J. Environ. Res. Public Health 2020, 17, 2179. [Google Scholar] [CrossRef]
- Ying, X.L.; Gao, Z.Y.; Yan, J.; Zhang, M.; Wang, J.; Xu, J.; Markowitz, M.; Yan, C.H. Sources, symptoms and characteristics of childhood lead poisoning: Experience from a lead specialty clinic in China. Clin. Toxicol. 2018, 56, 397–403. [Google Scholar] [CrossRef]
- Abdelli, N.; Peng, L.; Keping, C. Silkworm, Bombyx mori, as an alternative model organism in toxicological research. Environ. Sci. Pollut. Res. Int. 2018, 25, 35048–35054. [Google Scholar] [CrossRef]
- Ma, S.Y.; Smagghe, G.; Xia, Q.Y. Genome editing in Bombyx mori: New opportunities for silkworm functional genomics and the sericulture industry. Insect Sci. 2019, 26, 964–972. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, S.; Yan, X.; Qin, Z.; Jia, C.; Li, Z.; Zhang, J.; Huang, R. The mobility of cadmium and lead in the soil-mulberry-silkworm system. Chemosphere 2020, 242, 125179. [Google Scholar] [CrossRef]
- Wan, X.; Lei, M.; Chen, T.; Tan, Y.; Yang, J. Safe utilization of heavy-metal-contaminated farmland by mulberry tree cultivation and silk production. Sci. Total Environ. 2017, 599–600, 1867–1873. [Google Scholar] [CrossRef]
- Qian, H.; Li, G.; He, Q.; Zhang, H.; Xu, A. Analysis of differentially expressed genes between fluoride-sensitive and fluoride-endurable individuals in midgut of silkworm, Bombyx mori. Gene 2016, 588, 47–53. [Google Scholar] [CrossRef]
- Li, S.; Jiang, H.; Qiao, K.; Gui, W.; Zhu, G. Insights into the effect on silkworm (Bombyx mori) cocooning and its potential mechanisms following non-lethal dose tebuconazole exposure. Chemosphere 2019, 234, 338–345. [Google Scholar] [CrossRef]
- Borowska, J.; Pyza, E. Effects of heavy metals on insect immunocompetent cells. J. Insect Physiol. 2011, 57, 760–770. [Google Scholar] [CrossRef]
- Shu, Y.; Zhang, G.; Wang, J. Response of the common cutworm Spodoptera litura to zinc stress: Zn accumulation, metallothionein and cell ultrastructure of the midgut. Sci. Total Environ. 2012, 438, 210–217. [Google Scholar] [CrossRef]
- Shu, Y.; Zhou, J.; Lu, K.; Li, K.; Zhou, Q. Response of the common cutworm Spodoptera litura to lead stress: Changes in sex ratio, Pb accumulations, midgut cell ultrastructure. Chemosphere 2015, 139, 441–451. [Google Scholar] [CrossRef]
- Breton, J.; Le Clère, K.; Daniel, C.; Sauty, M.; Nakab, L.; Chassat, T.; Dewulf, J.; Penet, S.; Carnoy, C.; Thomas, P.; et al. Chronic ingestion of cadmium and lead alters the bioavailability of essential and heavy metals, gene expression pathways and genotoxicity in mouse intestine. Arch. Toxicol. 2013, 87, 1787–1795. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, Y.; Wang, S.; Han, S.; Liu, J. Lead in the soil-mulberry (Morus alba L.)-silkworm (Bombyx mori) food chain: Translocation and detoxification. Chemosphere 2015, 128, 171–177. [Google Scholar] [CrossRef]
- Liu, Y.; Xin, Z.Z.; Song, J.; Zhu, X.Y.; Liu, Q.N.; Zhang, D.Z.; Tang, B.P.; Zhou, C.L.; Dai, L.S. Transcriptome Analysis Reveals Potential Antioxidant Defense Mechanisms in Antheraea pernyi in Response to Zinc Stress. J. Agric. Food Chem. 2018, 66, 8132–8141. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury, Lead, Chromium, Cadmium, and arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef]
- Alengebawy, A.; Abdelkhalek, S.T.; Qureshi, S.R.; Wang, M.Q. Heavy Metals and Pesticides Toxicity in Agricultural Soil and Plants: Ecological Risks and Human Health Implications. Toxics 2021, 9, 42. [Google Scholar] [CrossRef]
- Li, F.; Hu, J.; Tian, J.; Xu, K.; Ni, M.; Wang, B.; Shen, W.; Li, B. Effects of phoxim on nutrient metabolism and insulin signaling pathway in silkworm midgut. Chemosphere 2016, 146, 478–485. [Google Scholar] [CrossRef]
- Wang, Y.H.; Gu, Z.Y.; Wang, J.M.; Sun, S.S.; Wang, B.B.; Jin, Y.Q.; Shen, W.D.; Li, B. Changes in the activity and the expression of detoxification enzymes in silkworms (Bombyx mori) after phoxim feeding. Pestic. Biochem. Physiol. 2013, 105, 13–17. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, J.; Qi, T.; Ge, B.; Wang, Z.; Jiang, S.; Liu, Q.; Zhang, H.; Ding, G.; Tang, B. Transcriptome Analysis of Hepatopancreas from the Cr (VI)-Stimulated Mantis Shrimp (Oratosquilla oratoria) by Illumina Paired-End Sequencing: Assembly, Annotation, and Expression Analysis. J. Agric. Food Chem. 2018, 66, 2598–2606. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. The Pfam protein families database. Nucleic Acids Res. 2007, 36, D281–D288. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Adesina, J.M. Antioxidant and detoxifying enzymes response of stored product insect pests to bioactive fractions of botanical extracts used as stored grains protectant. Ann. Environ. Sci. Toxicol. 2023, 7, 43–51. [Google Scholar] [CrossRef]
- Wang, X.R.; Wang, C.; Ban, F.X.; Zhu, D.T.; Liu, S.S.; Wang, X.W. Genome-wide identification and characterization of HSP gene superfamily in whitefly (Bemisia tabaci) and expression profiling analysis under temperature stress. Insect Sci. 2017, 26, 44–57. [Google Scholar] [CrossRef]
- Monchanin, C.; Blanc-Brude, A.; Drujont, E.; Negahi, M.M.; Pasquaretta, C.; Silvestre, J.; Baqué, D.; Elger, A.; Barron, A.B.; Devaud, J.M.; et al. Chronic exposure to trace lead impairs honey bee learning. Ecotoxicol. Environ. Saf. 2021, 212, 112008. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Yu, H.Z.; Deng, M.J.; Ma, Y.; Fei, D.Q.; Wang, J.; Li, Z.; Meng, Y.; Xu, J.P. Comparative transcriptome analysis reveals significant metabolic alterations in eri-silkworm (Samia cynthia ricini) haemolymph in response to 1-deoxynojirimycin. PLoS ONE 2018, 13, e0191080. [Google Scholar] [CrossRef]
- Zhang, T.; Huang, Y.; Chen, X.; Zheng, F.; Shen, Y.; Chen, G.; Ye, Q.; Chen, K.; Xiao, X.; Peng, Y. Tetraphenylethylene-based AIE nanoprobes for labeling lysosome by two-photon imaging in living cells. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2024, 306, 123630. [Google Scholar] [CrossRef]
- Muthukottiappan, P.; Winter, D. A proteomic view on lysosomes. Mol. Omics 2021, 17, 842–859. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef]
- Xia, Q.; Cheng, D.; Duan, J.; Wang, G.; Cheng, T.; Zha, X.; Liu, C.; Zhao, P.; Dai, F.; Zhang, Z.; et al. Microarray-based gene expression profiles in multiple tissues of the domesticated silkworm (Bombyx mori). Genome Biol. 2007, 8, R162. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Y.; Fu, Q.; Jia, L.; Xiang, Z.; He, N. Proteomic Analysis of Larval Midgut from the Silkworm (Bombyx mori). Comp. Funct. Genom. 2011, 2011, 876064. [Google Scholar] [CrossRef]
- Wang, R.L.; Li, J.; Staehelin, C.; Xin, X.W.; Su, Y.J.; Zeng, R.S. Expression Analysis of Two P450 Monooxygenase Genes of the Tobacco Cutworm Moth (Spodoptera litura) at Different Developmental Stages and in Response to Plant Allelochemicals. J. Chem. Ecol. 2015, 41, 111–119. [Google Scholar] [CrossRef]
- Guo, Y.; Wu, H.; Zhang, X.; Ma, E.; Guo, Y.; Zhu, K.Y.; Zhang, J. RNA interference of cytochrome P450 CYP6F subfamily genes affects susceptibility to different insecticides in Locusta migratoria. Pest Manag. Sci. 2016, 72, 2154–2165. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Montella, I.R.; Schama, R.; Valle, D. The classification of esterases: An important gene family involved in insecticide resistance—A Review. Mem. Inst. Oswaldo Cruz 2012, 107, 437–449. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free. Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, J.; Wu, K.; Chen, S.; Wang, J.; Shu, Y. Complex Cd–Pb Multigenerational Exposure Improves the Growth and Food Utilization of the Cutworm Spodoptera litura. Agronomy 2023, 13, 1207. [Google Scholar] [CrossRef]
- Mathew, B.B.; Krishnamurthy, N.B. Assessment of Lead Toxicity Using Drosophila melanogaster as a Model. J. Clin. Toxicol. 2018, 8, 380. [Google Scholar] [CrossRef]
- Gu, Z.Y.; Sun, S.S.; Wang, Y.H.; Wang, B.B.; Xie, Y.; Ma, L.; Wang, J.M.; Shen, W.D.; Li, B. Transcriptional characteristics of gene expression in the midgut of domestic silkworms (Bombyx mori) exposed to phoxim. Pestic. Biochem. Physiol. 2013, 105, 36–43. [Google Scholar] [CrossRef]
- Zhou, J.; Shu, Y.; Zhang, G.; Zhou, Q. Lead exposure improves the tolerance of Spodoptera litura (Lepidoptera: Noctuidae) to cypermethrin. Chemosphere 2012, 88, 507–513. [Google Scholar] [CrossRef]
- Chen, J.; Guo, Y.; Huang, S.; Zhan, H.; Zhang, M.; Wang, J.; Shu, Y. Integration of transcriptome and proteome reveals molecular mechanisms underlying stress responses of the cutworm, Spodoptera litura, exposed to different levels of lead (Pb). Chemosphere 2021, 283, 131205. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Y.; Shi, Y.-X.; Jiang, Q.; Jin, Y.; Chen, F.-X.; Tang, W.-H.; Peng, Q.; Liu, Q.-N.; Tang, B.-P.; Wang, J.-L. Transcriptome Analysis Reveals Antioxidant Defense Mechanisms in the Silkworm Bombyx mori after Exposure to Lead. Animals 2024, 14, 1822. https://doi.org/10.3390/ani14121822
Ye Y, Shi Y-X, Jiang Q, Jin Y, Chen F-X, Tang W-H, Peng Q, Liu Q-N, Tang B-P, Wang J-L. Transcriptome Analysis Reveals Antioxidant Defense Mechanisms in the Silkworm Bombyx mori after Exposure to Lead. Animals. 2024; 14(12):1822. https://doi.org/10.3390/ani14121822
Chicago/Turabian StyleYe, Yang, Yan-Xia Shi, Qi Jiang, Ye Jin, Fan-Xing Chen, Wen-Hui Tang, Qin Peng, Qiu-Ning Liu, Bo-Ping Tang, and Jia-Lian Wang. 2024. "Transcriptome Analysis Reveals Antioxidant Defense Mechanisms in the Silkworm Bombyx mori after Exposure to Lead" Animals 14, no. 12: 1822. https://doi.org/10.3390/ani14121822
APA StyleYe, Y., Shi, Y.-X., Jiang, Q., Jin, Y., Chen, F.-X., Tang, W.-H., Peng, Q., Liu, Q.-N., Tang, B.-P., & Wang, J.-L. (2024). Transcriptome Analysis Reveals Antioxidant Defense Mechanisms in the Silkworm Bombyx mori after Exposure to Lead. Animals, 14(12), 1822. https://doi.org/10.3390/ani14121822