
Long Non-Coding RNAs Differentially Expressed in Swine Fetuses


 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Declaration of Ethics
2.2. Animal Experiment
2.3. RNA Extraction and Library Preparation
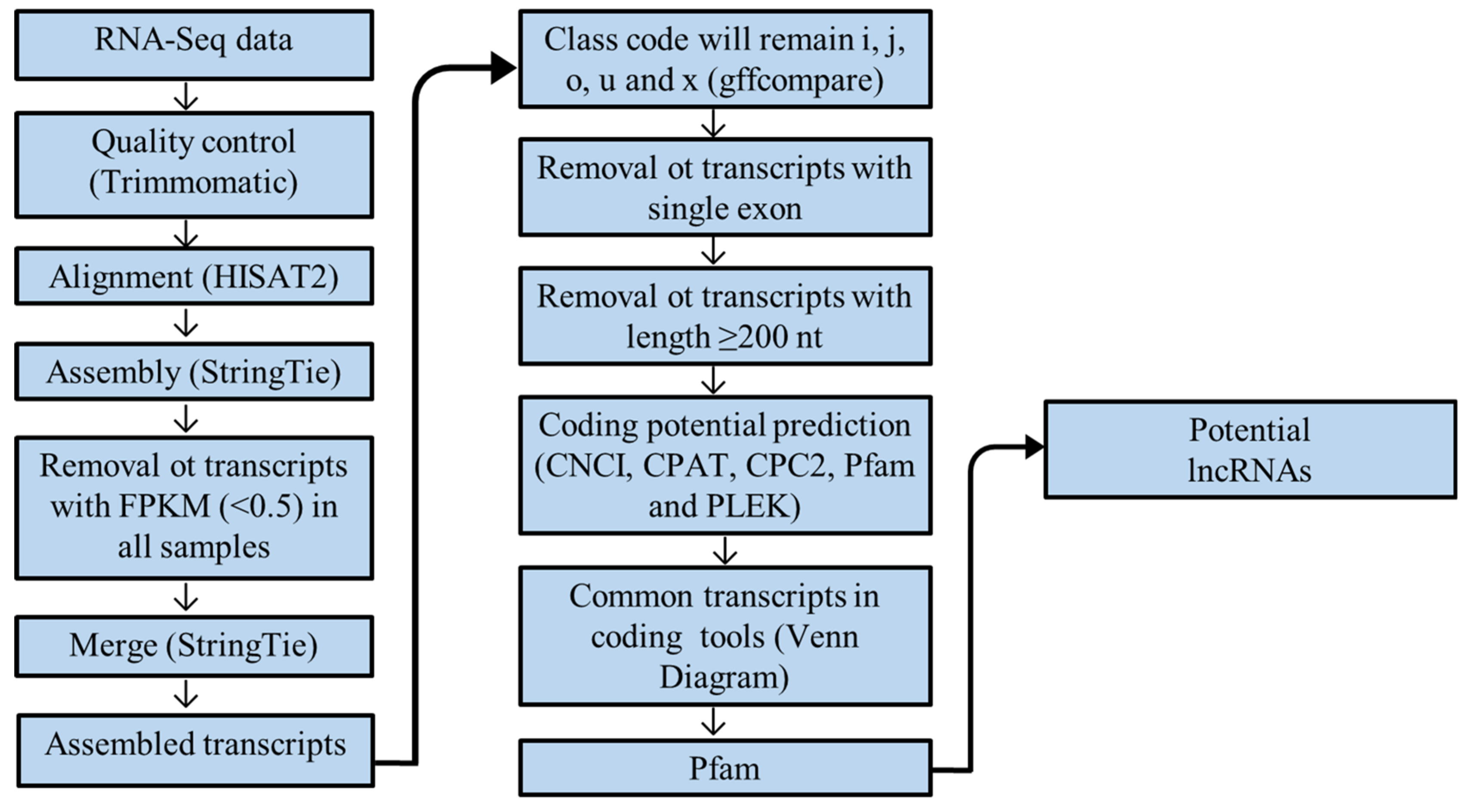
2.4. Quality Control, Mapping, and Transcriptome Assembly
2.5. Identification of Potentially New lncRNAs
2.6. Identification of Differentially Expressed (DE) lncRNAs
2.7. Target Gene Prediction and Functional Annotations
3. Results
3.1. Mapping and Assembly of Transcripts
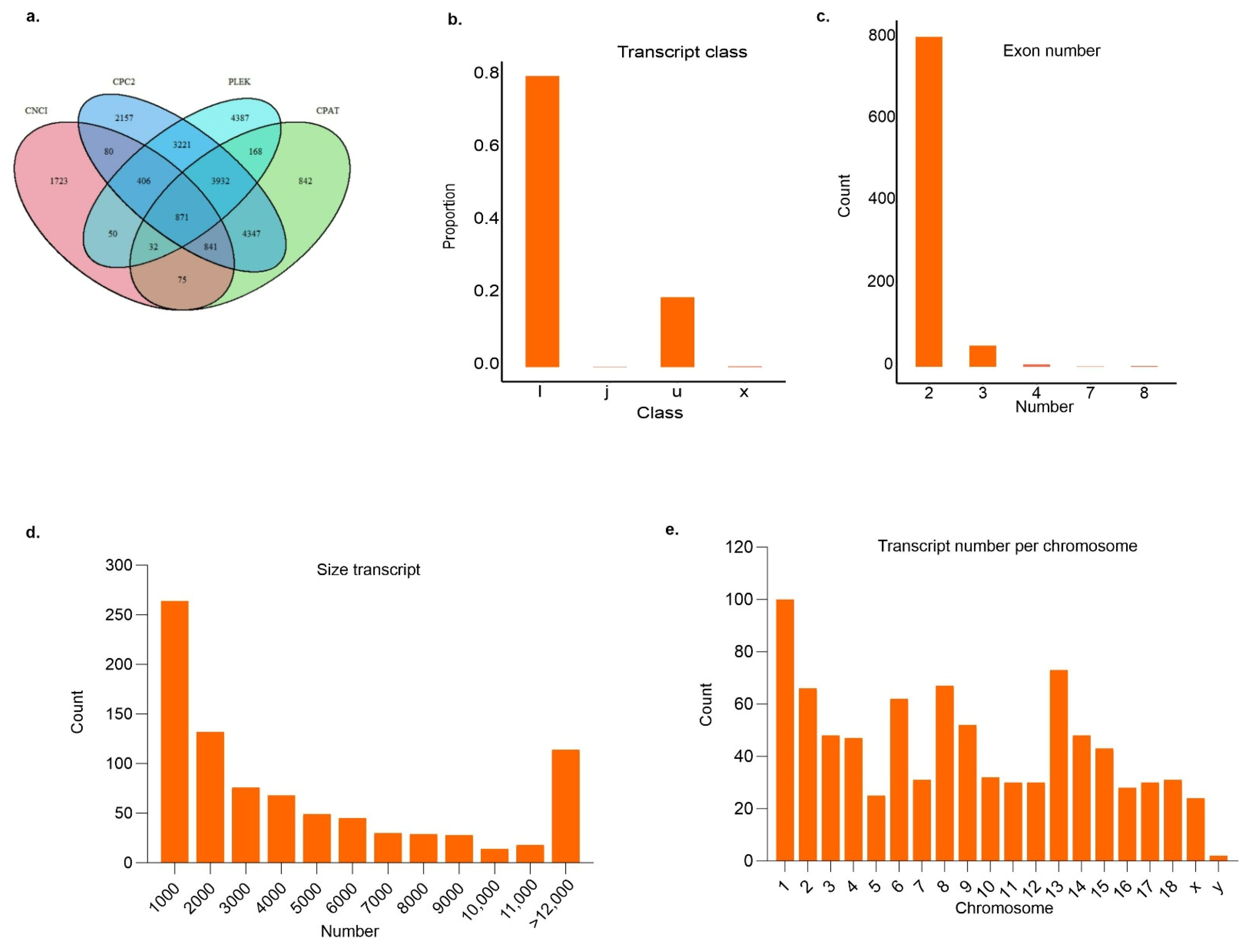
3.2. Identification and Characterization of Potentially New lncRNAs
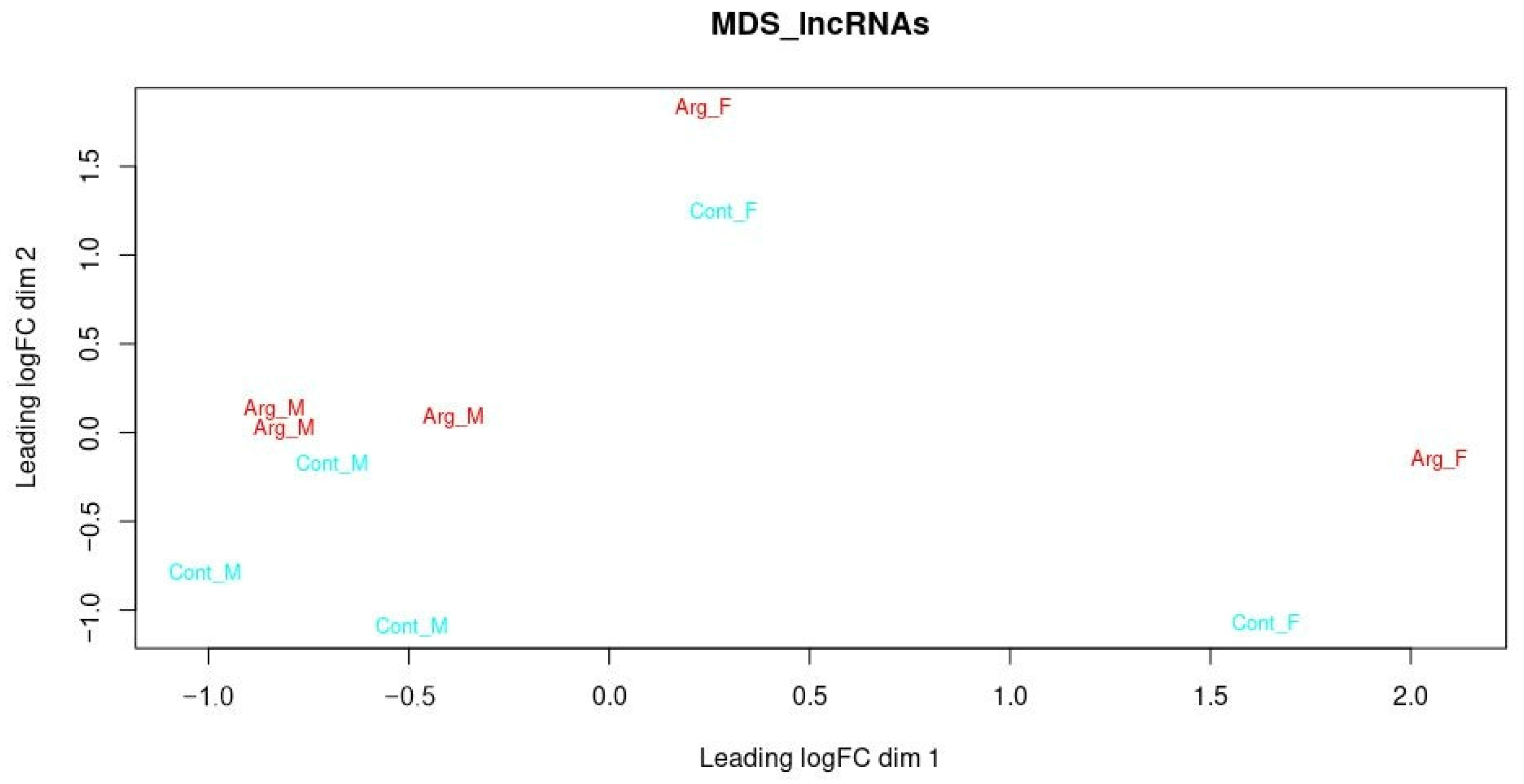
3.3. Differentially Expressed lncRNAs
3.4. Neighboring Genes of the DE lncRNAs
3.5. Potentially New DE lncRNAs Concerning Sex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, H.; Srikanth, K.; Park, W.; Lee, S.H.; Choi, B.H.; Kim, H.; Park, J.E. Transcriptome analysis to identify long non coding RNA (lncRNA) and characterize their functional role in back fat tissue of pig. Gene 2019, 703, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. BBA 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Sebastian-delaCruz, M.; Gonzalez-Moro, I.; Olazagoitia-Garmendia, A.; Castellanos-Rubio, A.; Santin, I. The role of lncRNA in gene expression regulation through mRNA stabilization. Non-Coding RNA 2021, 7, 3. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in development and disease: Background, mechanisms, and therapeutic approaches. Physiol. Ver. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [PubMed]
- Dykes, I.M.; Emanueli, C. Transcriptional and post-transcriptional gene regulation by long non-coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Liu, T.; Yousuf, S.; Zhang, X.; Huang, W.; Li, A.; Miao, X. Identification and analysis of lncRNA, miRNA and mRNA related to subcutaneous and intramuscular fat in Laiwu pigs. Front. Endocrinol. 2023, 13, 1081460. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, Y.; Huang, R.; Zhai, Y.; Wu, D.; An, X.; Li, Z. LncRNA affects epigenetic reprogramming of porcine embryo development by regulating global epigenetic modification and the downstream gene SIN3A. Front. Physiol. 2022, 13, 971965. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.X.; Liang, H.; Wu, Z.W.; Huo, L.J.; Du, Z.Q. Identification of lncRNAs involved in maternal-to-zygotic transition of in vitro-produced porcine embryos by single-cell RNA-seq. Reprod. Domest. Anim. 2022, 57, 111–122. [Google Scholar] [CrossRef]
- Wang, S.; Wang, H.; Liu, J.; Zhang, X.; Yang, Y.; Lu, C.; Gao, P. Expression patterns and functional analysis of porcine lnc-34015. Anim. Biotechnol. 2023, 34, 2251–2261. [Google Scholar] [CrossRef]
- Xu, C.; Shi, J.; Huang, R.; Wu, Z.; Wu, S.; Bao, W. Transcriptome-Wide lncRNA and mRNA Profiling of Spleens from Meishan Pigs at Different Development Stages. Animals 2022, 12, 2676. [Google Scholar] [CrossRef]
- Taylor, D.H.; Chu, E.T.J.; Spektor, R.; Soloway, P.D. Long non-coding RNA regulation of reproduction and development. Mol. Reprod. Dev. 2015, 82, 932–956. [Google Scholar] [CrossRef]
- Rastetter, R.H.; Smith, C.A.; Wilhelm, D. The role of non-coding RNAs in male sex determination and differentiation. Reproduction 2015, 150, R93–R107. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, R.; Sato, T.; Ogawa, T.; Okano, H.; Noce, T. A noncoding RNA containing a SINE-B1 motif associates with meiotic metaphase chromatin and has an indispensable function during spermatogenesis. PLoS ONE 2017, 12, e0179585. [Google Scholar] [CrossRef]
- Burgos, M.; Hurtado, A.; Jiménez, R.; Barrionuevo, F.J. Non-Coding RNAs: lncRNAs, miRNAs, and piRNAs in sexual development. Sex. Dev. 2021, 15, 335–350. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Zemel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Nature 1991, 351, 153–155. [Google Scholar] [CrossRef]
- Dinger, M.E.; Amaral, P.P.; Mercer, T.R.; Pang, K.C.; Bruce, S.J.; Gardiner, B.B.; Mattick, J.S. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008, 18, 1433–1445. [Google Scholar] [CrossRef]
- Moindrot, B.; Brockdorff, N. RNA binding proteins implicated in Xist-mediated chromosome silencing. Semin. Cell Dev. Biol. 2016, 56, 58–70. [Google Scholar] [CrossRef]
- Brownmiller, T.; Juric, J.A.; Ivey, A.D.; Harvey, B.M.; Westemeier, E.S.; Winters, M.T.; Martinez, I. Y chromosome lncRNA are involved in radiation response of male non–small cell lung cancer cells. Cancer Res. 2020, 80, 4046–4057. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Rajender, S. Long non-coding RNAs (lncRNAs) in spermatogenesis and male infertility. Reprod. Biol. Endocrinol. 2020, 18, 103. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Hou, J.; Liu, Y.; Liu, Y.; Yang, H.; Li, Z.; He, Z. The roles and regulation of Sertoli cells in fate determinations of spermatogonial stem cells and spermatogenesis. Sem. Cell Dev. Biol. 2014, 29, 66–75. [Google Scholar] [CrossRef]
- Zhang, M.; Li, N.; Liu, W.; Du, X.; Wei, Y.; Yang, D.; Hua, J. Eif2s3y promotes the proliferation of spermatogonial stem cells by activating ERK signaling. Stem Cells Internat. 2021, 2021, 6668658. [Google Scholar] [CrossRef]
- Johansson, M.M.; Pottmeier, P.; Suciu, P.; Ahmad, T.; Zaghlool, A.; Halvardson, J.; Jazin, E. Novel Y-chromosome long non-coding RNAs expressed in human male CNS during early development. Front. Genet. 2019, 10, 891. [Google Scholar] [CrossRef]
- Costa, K.A.; Saraiva, A.; Guimaraes, J.D.; Marques, D.B.D.; Machado-neves, M.; Reis, L.M.B.; Alberto, F.; Veroneze, R.; Oliveira, L.F.D.; Garcia, I.S.; et al. Dietary L-arginine supplementation during early gestation of gilts affects conceptuses development. Theriogenology 2019, 140, 62–71. [Google Scholar] [CrossRef]
- Teixeira, S.A.; Marques, D.B.D.; Costa, T.C.; Oliveira, H.C.; Costa, K.A.; Carrara, E.R.; Silva, W.D.; Guimarães, J.D.; Neves, M.M.; Ibelli, A.M.G.; et al. Transcription Landscape of the Early Developmental Biology in Pigs. Animals 2021, 11, 1443. [Google Scholar] [CrossRef]
- Teixeira, S.A.; Ibelli, A.M.G.; Cantão, E.; Oliveira, H.C.D.; Ledur, M.C.; Peixoto, J.d.O.; Marques, D.B.D.; Costa, K.A.; Coutinho, L.L.; Guimarães, S.E.F. Sex Determination Using RNA-Sequencing Analyses in Early Prenatal Pig Development. Genes 2019, 10, 1010. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Smyth, G.K.; Ritchie, M.; Thorne, N.; Wettenhall, J.; Shi, W.; Hu, Y. Limma: Linear Models for Microarray and RNA-Seq Data User’s Guide; The Walter and Eliza Hall Institute of Medical Research: Melbourne, Australia, 2002. [Google Scholar]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Dong, X.; Tian, L.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 2016, 5, 1408. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controllin the false discovery rate: A practical and powerful approch to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Yan, P.; Luo, S.; Lu, J.Y.; Shen, X. Cis- and trans-acting lncRNAs in pluripotency and reprogramming. Curr. Opin. Genet. Dev. 2017, 46, 170–178. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional classification and experimental dissection of long noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Chen, H.; Palmer, J.S.; Thiagarajan, R.D.; Dinger, M.E.; Lesieur, E.; Chiu, H.; Wilhelm, D. Identification of novel markers of mouse fetal ovary development. PLoS ONE 2012, 7, e41683. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Li, W.; Tian, H.; Hu, T.; Wang, L.; Lin, Y.; Sun, F. Sequential expression of long noncoding RNA as mRNA gene expression in specific stages of mouse spermatogenesis. Sci. Rep. 2014, 4, 5966. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Chang, H.Y. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, I.A.; Dundr, M. Chromatin loops and causality loops: The influence of RNA upon spatial nuclear architecture. Chromosoma 2017, 126, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; Cooper, P.; Smith, S.; McCabe, V.M.; Norris, D.P.; Penny, G.D.; Rastan, S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 1991, 351, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N. Localized accumulation of Xist RNA in X chromosome inactivation. Open Biol. 2019, 9, 190213. [Google Scholar] [CrossRef] [PubMed]
- Jégu, T.; Aeby, E.; Lee, J.T. The X chromosome in space. Nat. Revi. Genet. 2017, 18, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Vallot, C.; Huret, C.; Lesecque, Y.; Resch, A.; Oudrhiri, N.; Bennaceur-Griscelli, A.; Rougeulle, C. XACT, a long noncoding transcript coating the active X chromosome in human pluripotent cells. Nat. Genet. 2013, 45, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, Q.; Hua, L.; Chen, J.; Zhang, J.; Bai, H.; Bai, X. Comprehensive analysis of differentially expressed mRNA, lncRNA and circRNA and their ceRNA networks in the longissimus dorsi muscle of two different pig breeds. Int. J. Mol. Sci. 2019, 20, 1107. [Google Scholar] [CrossRef]
- Vakilian, H.; Mirzaei, M.; Sharifi Tabar, M.; Pooyan, P.; Habibi Rezaee, L.; Parker, L.; Salekdeh, G.H. DDX3Y, a male-specific region of Y chromosome gene, may modulate neuronal differentiation. J. Proteome Res. 2015, 14, 3474–3483. [Google Scholar] [CrossRef]
- Jaroszynski, L.; Zimmer, J.; Fietz, D.; Bergmann, M.; Kliesch, S.; Vogt, P.H. Translational control of the AZFa gene DDX3Y by 5′ UTR exon-T extension. Int. J. Androl. 2011, 34, 313–326. [Google Scholar] [CrossRef]
- Rauschendorf, M.A.; Zimmer, J.; Hanstein, R.; Dickemann, C.; Vogt, P.H. Complex transcriptional control of the AZFa gene DDX3Y in human testis. Int. J. Androl. 2011, 34, 84–96. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Piao, F.; Wang, X. Subchronic exposure to arsenic inhibits spermatogenesis and downregulates the expression of ddx3y in testis and epididymis of mice. Toxicol. Sci. 2012, 128, 482–489. [Google Scholar] [CrossRef]
- Blair, L.P.; Cao, J.; Zou, M.R.; Sayegh, J.; Yan, Q. Epigenetic regulation by lysine demethylase 5 (KDM5) enzymes in cancer. Cancers 2011, 3, 1383–1404. [Google Scholar] [CrossRef]
- Merten, M.; Greiner, J.F.W.; Niemann, T.; Grosse Venhaus, M.; Kronenberg, D.; Stange, R.; Wähnert, D.; Kaltschmidt, C.; Vordemvenne, T.; Kaltschmidt, B. Human Sex Matters: Y-Linked Lysine Demethylase 5D Drives Accelerated Male Craniofacial Osteogenic Differentiation. Cells 2022, 11, 823. [Google Scholar] [CrossRef]
- Mizukami, H.; Kim, J.D.; Tabara, S.; Lu, W.; Kwon, C.; Nakashima, M.; Fukamizu, A. KDM5D-mediated H3K4 demethylation is required for sexually dimorphic gene expression in mouse embryonic fibroblasts. J. Biochem. 2019, 165, 335–342. [Google Scholar] [CrossRef]
- Fontanesi, L.; Scotti, E.; Russo, V. Differences in porcine amelogenin X and Y chromosome genes (AMELX and AMELY) and their application in sex determination in pigs. Mol. Reprod. Dev. 2008, 75, 1662–1668. [Google Scholar] [CrossRef]
- Decarpentrie, F.; Vernet, N.; Mahadevaiah, S.K.; Longepied, G.; Streichemberger, E.; Aknin-Seifer, I.; Mitchell, M.J. Human and mouse ZFY genes produce a conserved testis-specific transcript encoding a zinc finger protein with a short acidic domain and modified transactivation potential. Hum. Mol. Genet. 2012, 21, 2631–2645. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Metzler-Guillemain, C.; Toure, A.; Coutton, C.; Arnoult, C.; Ray, P.F. Single gene defects leading to sperm quantitative anomalies. Clin. Genet. 2017, 91, 208–216. [Google Scholar] [CrossRef]
- Wittkopp, P.J.; Haerum, B.K.; Clark, A.G. Evolutionary changes in cis and trans gene regulation. Nature 2004, 430, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Liu, T.; Fu, H.; Li, W.; Zheng, X. Genome-wide analysis of lncRNA stability in human. PLoS Comput. Biol. 2021, 17, e1008918. [Google Scholar] [CrossRef] [PubMed]
- Oron, E.; Mannervik, M.; Rencus, S.; Harari-Steinberg, O.; Neuman-Silberberg, S.; Segal, D.; Chamovitz, D.A. COP9 signalosome subunits 4 and 5 regulate multiple pleiotropic pathways in Drosophila melanogaster. Development 2002, 129, 4399–4409. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Wunder, J.S.; Gokgoz, N.; Gill, M.; Eskandarian, S.; Parkes, R.K.; Andrulis, I.L. COPS3 amplification and clinical outcome in osteosarcoma. Cancer 2007, 109, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lang, G.; Ito, S.; Bonnet, J.; Metzger, E.; Sawatsubashi, S.; Devys, D. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol. Cell 2008, 29, 92–101. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Varthi, M.; Sykes, S.M.; Phillips, C.; Warzecha, C.; Zhu, W.; McMahon, S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell 2008, 29, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Duan, B.; Sun, S.; Cui, J.; Du, J.; Zhang, Y. Folliculin interacts with Rab35 to regulate EGF-induced EGFR degradation. Front. Pharmacol. 2017, 8, 688. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhang, B.; Zhu, W.; Zhu, C.; Chen, L.; Zhao, Y.; Wang, Y.; Zhang, Y.; Riaz, A.; Tang, B.; et al. Expression of Phospholipase D Family Member 6 in Bovine Testes and Its Molecular Characteristics. Int. J. Mol. Sci. 2023, 24, 12172. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Chuma, S.; Yamamoto, Y.; Kuramochi-Miyagawa, S.; Totoki, Y.; Toyoda, A.; Sasaki, H. MITOPLD is a mitochondrial protein essential for nuage formation and piRNA biogenesis in the mouse germline. Dev. Cell 2011, 20, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Vallenius, T.; Vaahtomeri, K.; Kovac, B.; Osiceanu, A.M.; Viljanen, M.; Mäkelä, T.P. An association between NUAK2 and MRIP reveals a novel mechanism for regulation of actin stress fibers. J. Cell Sci. 2011, 124, 384–393. [Google Scholar] [CrossRef]
- Mulder, J.; Ariaens, A.; van den Boomen, D.; Moolenaar, W.H. p116Rip targets myosin phosphatase to the actin cytoskeleton and is essential for RhoA/ROCK-regulated neuritogenesis. Mol. Biol. Cell 2004, 15, 5516–5527. [Google Scholar] [CrossRef]
- Mulder, J.; Ariaens, A.; van Horck, F.P.; Moolenaar, W.H. Inhibition of RhoA-mediated SRF activation by p116Rip. FEBS Lett. 2005, 579, 6121–6127. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Sample | Mappead_Reads | Mapping (%) |
|---|---|---|
| 8209_E1 | 25,445,184 | 85.26 |
| 8218_E1 | 27,694,122 | 83.57 |
| 8218_E5 | 27,209,218 | 83.57 |
| 8219_E1 | 32,010,420 | 83.95 |
| 8219_E2 | 27,031,814 | 84.01 |
| 8219_E5 | 23,178,930 | 82.75 |
| 8273_E1 | 23,651,782 | 84.14 |
| 8274_E1 | 22,753,549 | 84.46 |
| 8274_E5 | 24,454,713 | 83.09 |
| 8284_E5 | 20,739,162 | 83.28 |
| Ensembl ID | logFC 1 | adj.P.Val 2 | Strand |
|---|---|---|---|
| ENSSSCG00000053329 | 6.737 | 7.14 × 10−6 | Forward |
| ENSSSCG00000049587 | 6.575 | 1.28 × 10−5 | Forward |
| ENSSSCG00000044223 | 5.490 | 5.78 × 10−5 | Reverse |
| ENSSSCG00000044023 | 5.327 | 0.000242 | Forward |
| ENSSSCG00000057564 | 5.117 | 0.000242 | Forward |
| ENSSSCG00000043846 | 4.985 | 0.00036 | Reverse |
| ENSSSCG00000055301 | 4.347 | 0.006892 | Forward |
| MSTRG.22328.1 | 5.938 | 2.12 × 10−5 | Reverse |
| lncRNAs | Gene Upstream | Gene Downstream |
|---|---|---|
| ENSSSCG00000053329 | LOC100626157, ENSSSCG00000025253 | DDX3Y |
| ENSSSCG00000049587 | KDM5D | LOC110257970, LOC110257894 |
| ENSSSCG00000044223 | LOC100624149, ENSSSCG00000052941, EIF1AY, LOC102162178 | ZFY, AMELY |
| ENSSSCG00000044023 | ZFY, LOC100624149, ENSSSCG00000052941, EIF1AY | AMELY, ENSSSCG00000039365 |
| ENSSSCG00000057564 | ENSSSCG00000025253 | ENSSSCG00000026430, ENSSSCG00000027046 |
| ENSSSCG00000043846 | ENSSSCG00000025253 | DDX3Y, LOC100625207 |
| ENSSSCG00000055301 | COPS3, FLCN, PLD6, MPRIP, ENSSSCG00000040134 and USP22 | DHRS7B, TMEM11, NATD1, MAP2K3, ENSSSCG00000051348 and ENSSSCG00000034773 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, F.G.; Ibelli, A.M.G.; Cantão, M.E.; Oliveira, H.C.; Peixoto, J.O.; Ledur, M.C.; Guimarães, S.E.F. Long Non-Coding RNAs Differentially Expressed in Swine Fetuses. Animals 2024, 14, 1897. https://doi.org/10.3390/ani14131897
Campos FG, Ibelli AMG, Cantão ME, Oliveira HC, Peixoto JO, Ledur MC, Guimarães SEF. Long Non-Coding RNAs Differentially Expressed in Swine Fetuses. Animals. 2024; 14(13):1897. https://doi.org/10.3390/ani14131897
Chicago/Turabian StyleCampos, Francelly G., Adriana M. G. Ibelli, Maurício E. Cantão, Haniel C. Oliveira, Jane O. Peixoto, Mônica C. Ledur, and Simone E. F. Guimarães. 2024. "Long Non-Coding RNAs Differentially Expressed in Swine Fetuses" Animals 14, no. 13: 1897. https://doi.org/10.3390/ani14131897
APA StyleCampos, F. G., Ibelli, A. M. G., Cantão, M. E., Oliveira, H. C., Peixoto, J. O., Ledur, M. C., & Guimarães, S. E. F. (2024). Long Non-Coding RNAs Differentially Expressed in Swine Fetuses. Animals, 14(13), 1897. https://doi.org/10.3390/ani14131897