

Multi-Allelic Mitochondrial DNA Deletions in an Adult Dog with Chronic Weakness, Exercise Intolerance and Lactic Acidemia
 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Case
2.2. Histopathology, Histochemistry, Immunohistochemistry and Electron Microscopy
2.3. Whole Genome Sequencing and Variant Analysis
3. Results
3.1. Clinical Case
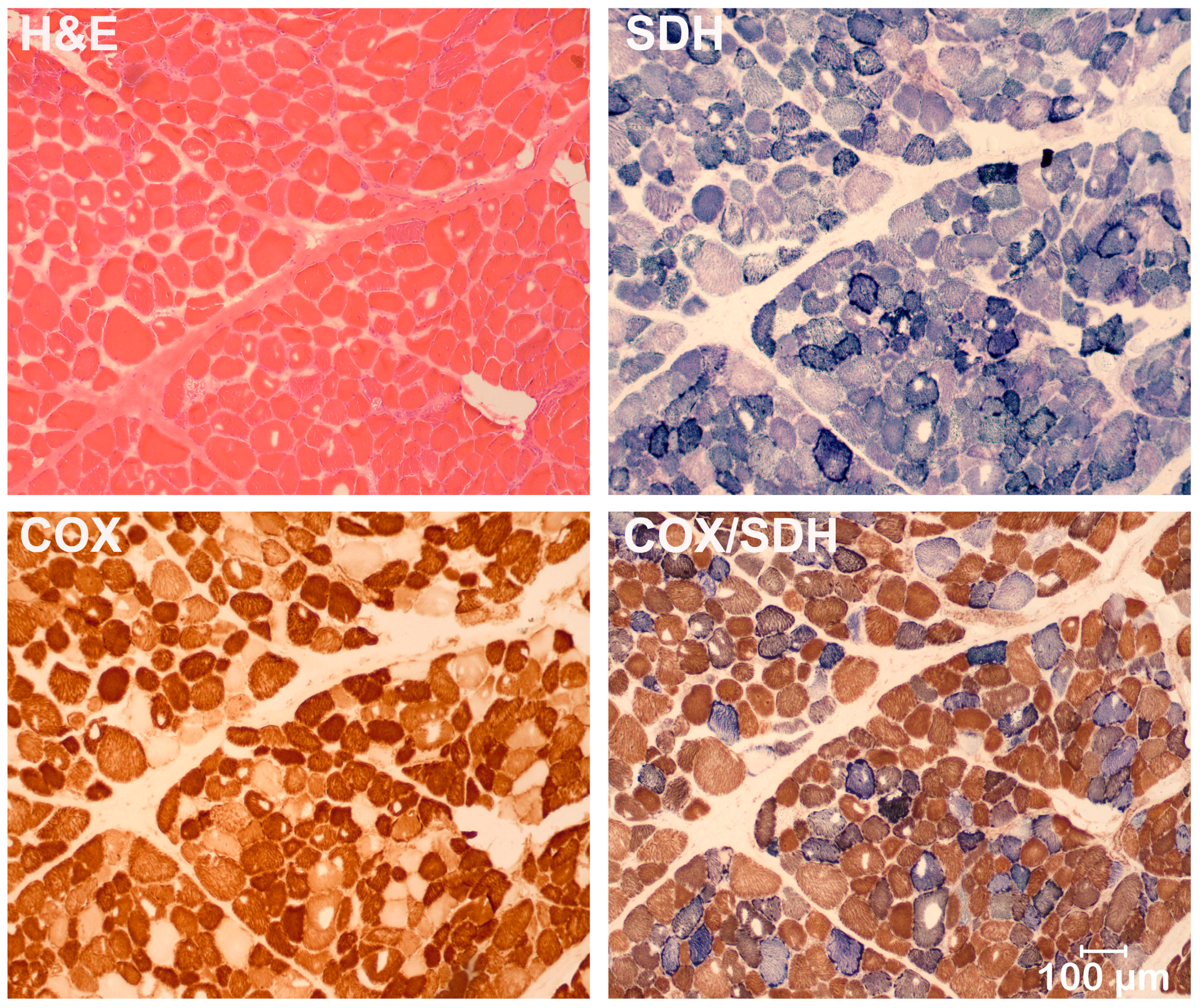
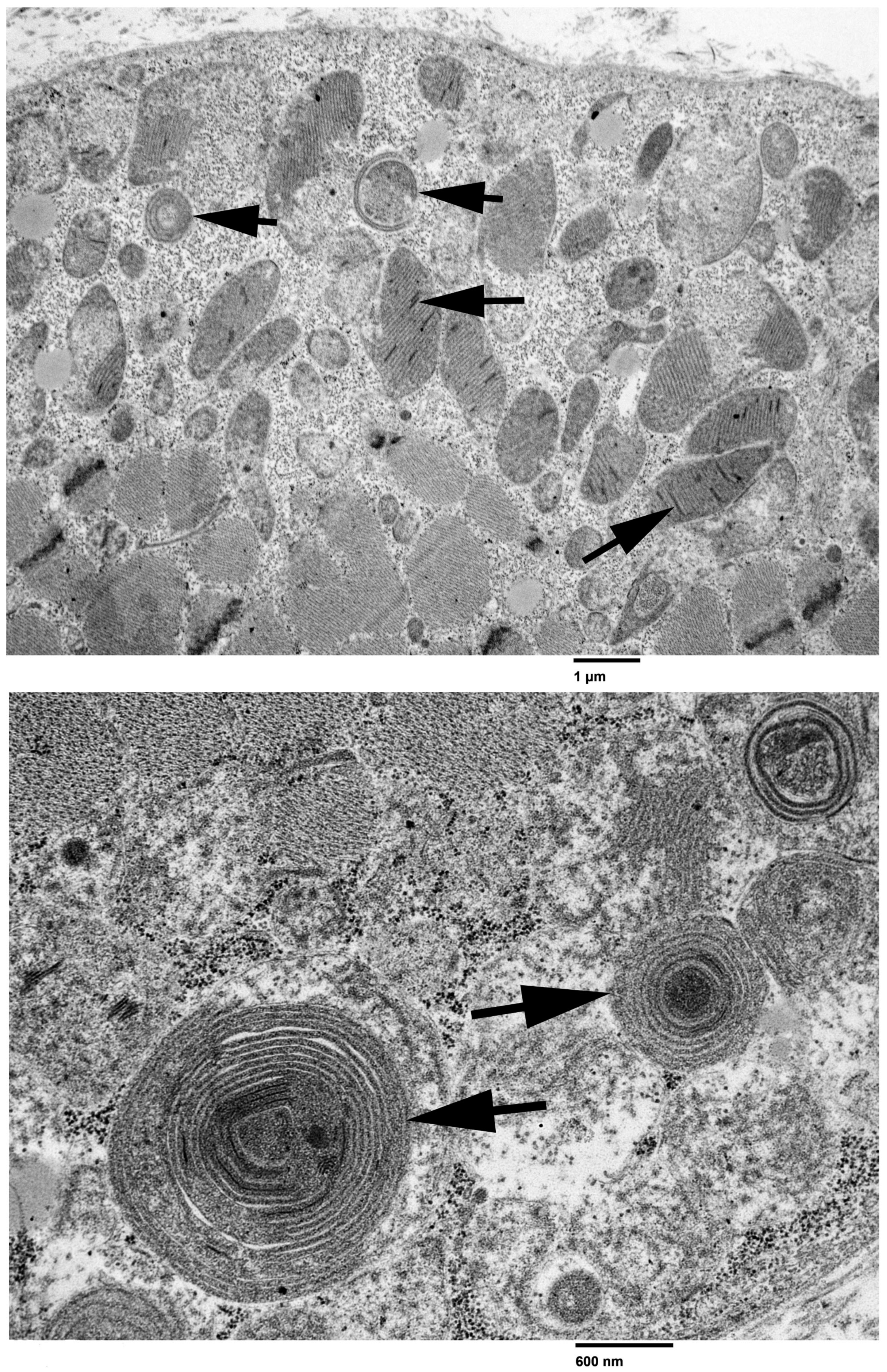
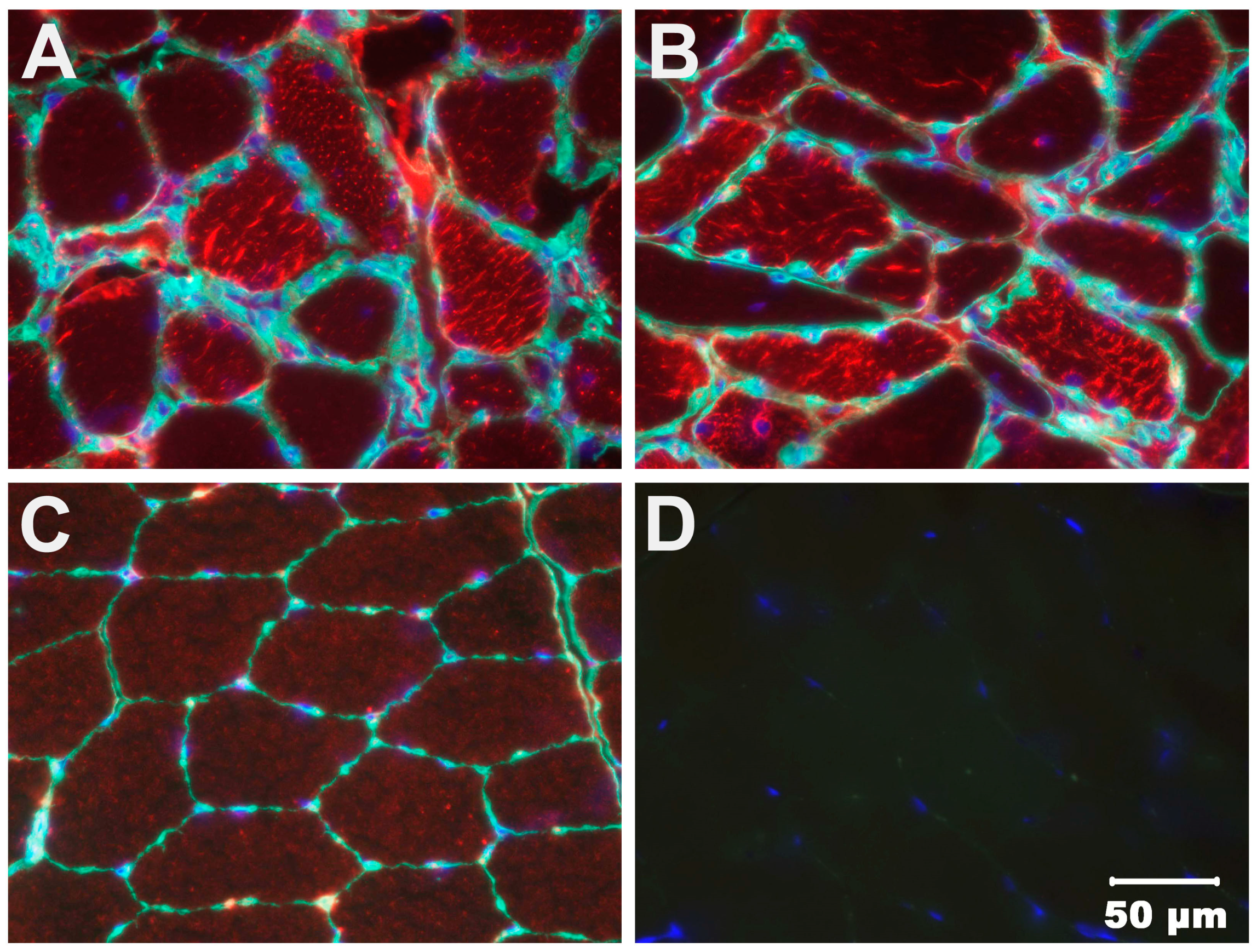
3.2. Light and Electron Microscopy
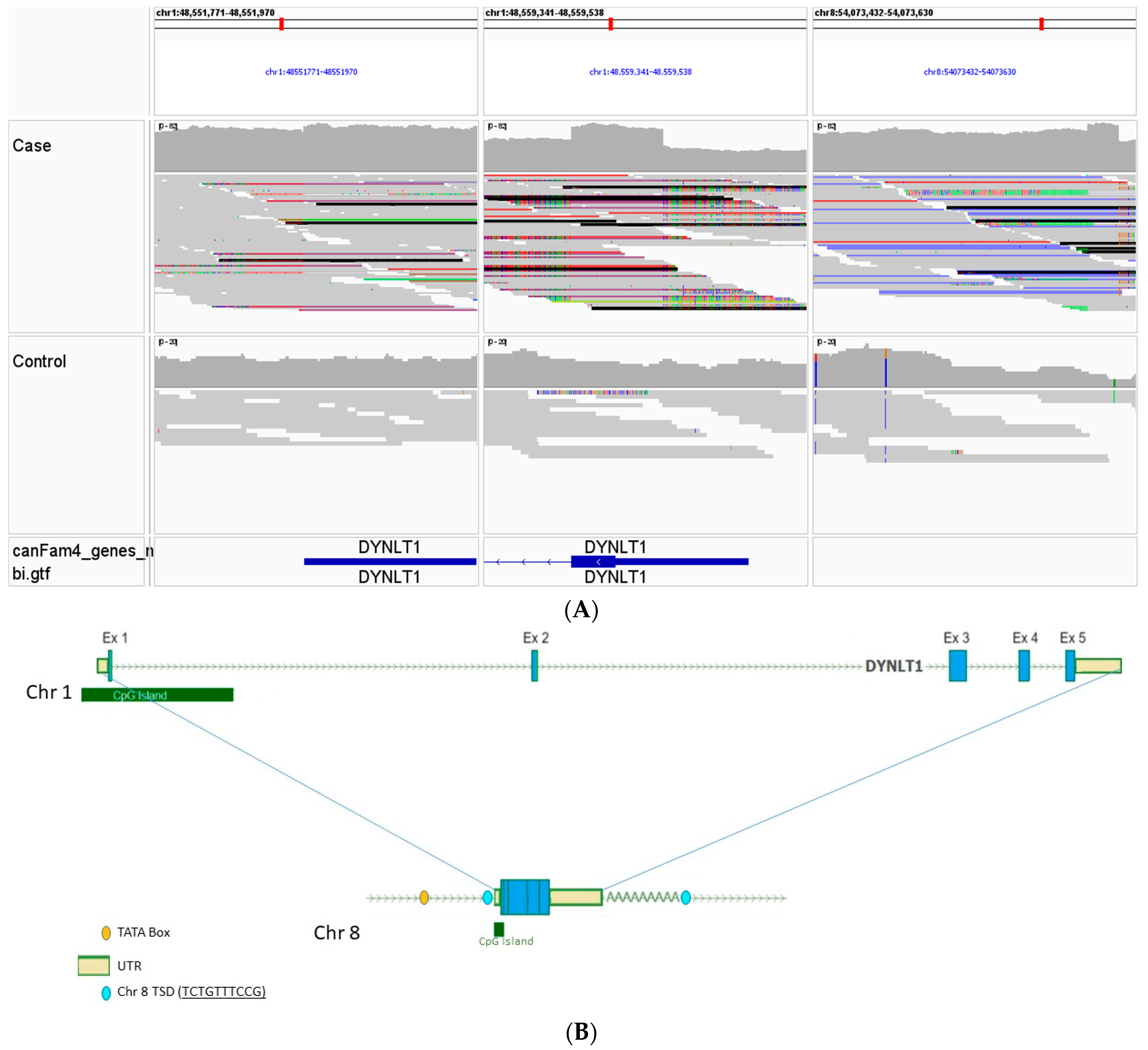
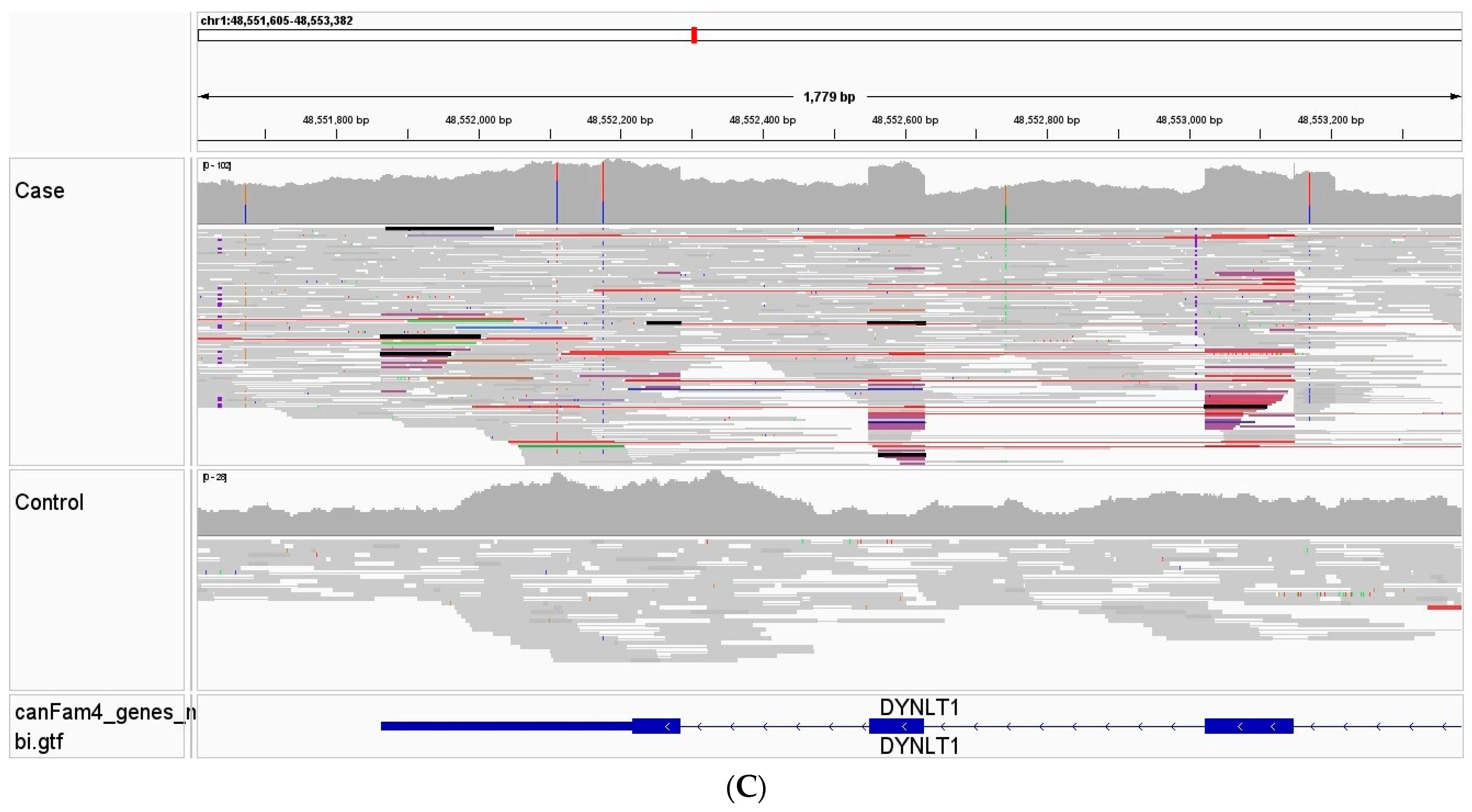
3.3. Whole Genome, Mitochondrial DNA Sequencing, and Variant Analysis
3.4. Immunohistochemical Staining
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.T.; Craven, L.; Russell, O.M.; Turnbull, D.M.; Vincent, A.E. Diagnosis and treatment of mitochondrial myopathies. Neurotherapeutics 2018, 15, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Emmanuele, V.; Ganesh, J.; Vladutiu, G.; Haas, R.; Kerr, D.; Saneto, R.P.; Cohen, B.H.; Van Hove, J.L.K.; Scaglia, F.; Hoppel, C.; et al. North American Mitochondrial Disease Consortium (NAMDC). Time to harmonize mitochondrial syndrome nomenclature and classification: A consensus from the North American Mitochondrial Disease Consortium (NAMDC). Mol. Genet. Metab. 2022, 136, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, E.A. Nuclear genetic defects of oxidative phosphorylation. Hum. Mol. Genet. 2001, 10, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Di Donato, S. Mitochondrial disorders. Brain 2004, 127, 2153–2172. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef]
- Zeviani, M.; Moraes, C.T.; DiMauro, S.; Nakase, H.; Bonilla, E.; Schon, E.A.; Rowland, L.P. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1988, 38, 1339–1346. [Google Scholar] [CrossRef]
- Moraes, C.T.; DiMauro, S.; Zeviani, M.; Lombes, A.; Shanske, S.; Miranda, A.F.; Nakase, H.; Bonilla, E.; Werneck, L.C.; Servidei, S.; et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N. Engl. J. Med. 1989, 320, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Santorelli, F.M.; Tanji, K.; Kulikova, R.; Shanske, S.; Vilarinho, L.; Hays, A.P.; DiMauro, S. Identification of a novel mutation in the mtDNA ND5 gene associated with MELAS. Biochem. Biophys. Res. Commun. 1997, 238, 326–328. [Google Scholar] [CrossRef]
- Kirby, D.M.; McFarland, R.; Ohtake, A.; Dunning, C.; Ryan, M.T.; Wilson, C.; Ketteridge, D.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Vernau, K.M.; Runstadler, J.A.; Brown, E.A.; Cameron, J.M.; Huson, H.J.; Higgins, R.J.; Ackerley, C.; Sturges, B.K.; Dickinson, P.J.; Puschner, B.; et al. Genome-wide association analysis identifies a mutation in the thiamine transporter 2 (SLC19A3) gene associated with Alaskan Husky encephalopathy. PLoS ONE 2013, 8, e57195. [Google Scholar] [CrossRef]
- Drögemüller, M.; Letko, A.; Matiasek, K.; Jagannathan, V.; Corlazzoli, D.; Rosati, M.; Jurina, K.; Medl, S.; Gödde, T.; Rupp, S.; et al. SLC19A3 Loss-of-Function Variant in Yorkshire Terriers with Leigh-Like Subacute Necrotizing Encephalopathy. Genes 2020, 11, 1215. [Google Scholar] [CrossRef]
- Li, F.Y.; Cuddon, P.A.; Song, J.; Wood, S.L.; Patterson, J.S.; Shelton, G.D.; Duncan, I.D. Canine spongiform leukoencephalomyelopathy is associated with a missense mutation in cytochrome b. Neurobiol. Dis. 2006, 21, 35–42. [Google Scholar] [CrossRef]
- Christen, M.; Gregor, A.; Gutierrez-Quintana, R.; Bongers, J.; Rupp, A.; Penderis, J.; Shelton, G.D.; Jagannathan, V.; Zweier, C.; Leeb, T. NDUFS7 variant in dogs with Leigh syndrome and its functional validation in a Drosophila melanogaster model. Sci. Rep. 2024, 14, 2975. [Google Scholar] [CrossRef]
- Cameron, J.M.; Maj, M.C.; Levandovskiy, V.; MacKay, N.; Shelton, G.D.; Robinson, B.H. Identification of a canine model of pyruvate dehydrogenase phosphatase 1 deficiency. Mol. Genet. Metab. 2007, 90, 15–23. [Google Scholar] [CrossRef]
- Shelton, G.D.; Minor, K.M.; Li, K.; Naviaux, J.C.; Monk, J.; Wang, L.; Guzik, E.; Guo, L.T.; Porcelli, V.; Gorgoglione, R.; et al. A Mutation in the Mitochondrial Aspartate/Glutamate Carrier Leads to a More Oxidizing Intramitochondrial Environment and an Inflammatory Myopathy in Dutch Shepherd Dogs. J. Neuromuscul. Dis. 2019, 6, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Lepori, V.; Mühlhause, F.; Sewell, A.C.; Jagannathan, V.; Janzen, N.; Rosati, M.; Alves de Sousa, F.M.M.; Tschopp, A.; Schüpbach, G.; Matiasek, K.; et al. A Nonsense Variant in the ACADVL Gene in German Hunting Terriers with Exercise Induced Metabolic Myopathy. G3 2018, 8, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Vijayasarathy, C.; Giger, U.; Prociuk, U.; Patterson, D.F.; Breitschwerdt, E.B.; Avadhani, N.G. Canine mitochondrial myopathy associated with reduced mitochondrial mRNA and altered cytochrome c oxidase activities in fibroblasts and skeletal muscle. Comp. Biochem. Physiol. A Physiol. 1994, 109, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Tkaczyk-Wlizło, A.; Kowal, K.; Ślaska, B. Mitochondrial DNA alterations in the domestic dog (Canis lupus familiaris) and their association with development of diseases: A review. Mitochondrion 2022, 63, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Baranowska, I.; Jäderlund, K.H.; Nennesmo, I.; Holmqvist, E.; Heidrich, N.; Larsson, N.G.; Andersson, G.; Wagner, E.G.; Hedhammar, A.; Wibom, R.; et al. Sensory ataxic neuropathy in golden retriever dogs is caused by a deletion in the mitochondrial tRNATyr gene. PLoS Genet. 2009, 5, e1000499. [Google Scholar] [CrossRef] [PubMed]
- Meurs, K.M.; Friedenberg, S.G.; Olby, N.J.; Condit, J.; Weidman, J.; Rosenthal, S.; Shelton, G.D. A QIL1 Variant Associated with Ventricular Arrhythmias and Sudden Cardiac Death in the Juvenile Rhodesian Ridgeback Dog. Genes 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, V.; Sewry, C.A.; Oldfors, A. Histological and histochemical stains and reactions. In Muscle Biopsy: A Practical Approach, 5th ed; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Wang, C.; Wallerman, O.; Arendt, M.L.; Sundström, E.; Karlsson, Å.; Nordin, J.; Mäkeläinen, S.; Pielberg, G.R.; Hanson, J.; Ohlsson, Å.; et al. A novel canine reference genome resolves genomic architecture and uncovers transcript complexity. Commun. Biol. 2021, 4, 185. [Google Scholar] [CrossRef] [PubMed]
- Meadows, J.R.S.; Kidd, J.M.; Wang, G.D.; Parker, H.G.; Schall, P.Z.; Bianchi, M.; Christmas, M.J.; Bougiouri, K.; Buckley, R.M.; Hitte, C.; et al. Genome sequencing of 2000, canids by the Dog10K consortium advances the understanding of demography, genome function and architecture. Genome Biol. 2023, 24, 187. [Google Scholar] [CrossRef]
- Cullen, J.N.; Friedenberg, S.G. Whole Animal Genome Sequencing: User-friendly, rapid, containerized pipelines for processing, variant discovery, and annotation of short-read whole genome sequencing data. G3 2023, 13, jkad117. [Google Scholar] [CrossRef]
- Knudsen, S. Promoter2.0: For the recognition of PolII promoter sequences. Bioinformatics 1999, 15, 356–361. [Google Scholar] [CrossRef]
- Solovyev, V.V.; Shahmuradov, I.A.; Salamov, A.A. Identification of promoter regions and regulatory sites. Methods Mol. Biol. 2010, 674, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, P.J.; LeCouteur, R.A. Muscle and nerve biopsy. Vet. Clin. N. Am. Small Anim. Pract. 2002, 32, 63–102. [Google Scholar] [CrossRef] [PubMed]
- Glass, E.N.; Kent, M. The clinical examination for neuromuscular disease. Vet. Clin. N. Am. Small Anim. Pract. 2002, 32, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Picard, M.; Turnbull, D.M. The ageing neuromuscular system and sarcopenia: A mitochondrial perspective. J. Physiol. 2016, 594, 4499–4512. [Google Scholar] [CrossRef] [PubMed]
- Volpi, L.; Ricci, G.; Orsucci, D.; Alessi, R.; Bertolucci, F.; Piazza, S.; Simoncini, C.; Mancuso, M.; Siciliano, G. Metabolic myopathies: Functional evaluation by different exercise testing approaches. Musculoskelet. Surg. 2011, 95, 59–67. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Varughese, J.T.; Buchanan, S.K.; Pitt, A.S. The Role of Voltage-Dependent Anion Channel in Mitochondrial Dysfunction and Human Disease. Cells 2021, 10, 1737. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.d.; Xu, X.; Dang, Y.m.; Zhang, Y.m.; Zhang, J.p.; Hu, J.-Y.; Zhang, Q.; Dai, X.; Teng, M.; Zhang, D.-X.; et al. MAP4 Mechanism that Stabilizes Mitochondrial Permeability Transition in Hypoxia: Microtubule Enhancement and DYNLT1 Interaction with VDAC1. PLoS ONE 2011, 6, e28052. [Google Scholar] [CrossRef]
- Huang, L.; Wei, B.; Zhao, Y.; Gong, X.; Chen, L. DYNLT1 promotes mitochondrial metabolism to fuel breast cancer development by inhibiting ubiquitination degradation of VDAC1. Mol. Med. 2023, 29, 72. [Google Scholar] [CrossRef]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Naghdi, S.; Hajnóczky, G. VDAC2-specific cellular functions and the underlying structure. Biochim. Biophys. Acta 2016, 1863, 2503–2514. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef]
- Parker, H.G.; VonHoldt, B.M.; Quignon, P.; Margulies, E.H.; Shao, S.; Mosher, D.S.; Spady, T.C.; Elkahloun, A.; Cargill, M.; Jones, P.G.; et al. An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs. Science 2009, 325, 995–998. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shelton, G.D.; Mickelson, J.R.; Friedenberg, S.G.; Cullen, J.N.; Mehra, J.M.; Guo, L.T.; Minor, K.M. Multi-Allelic Mitochondrial DNA Deletions in an Adult Dog with Chronic Weakness, Exercise Intolerance and Lactic Acidemia. Animals 2024, 14, 1946. https://doi.org/10.3390/ani14131946
Shelton GD, Mickelson JR, Friedenberg SG, Cullen JN, Mehra JM, Guo LT, Minor KM. Multi-Allelic Mitochondrial DNA Deletions in an Adult Dog with Chronic Weakness, Exercise Intolerance and Lactic Acidemia. Animals. 2024; 14(13):1946. https://doi.org/10.3390/ani14131946
Chicago/Turabian StyleShelton, G. Diane, James R. Mickelson, Steven G. Friedenberg, Jonah N. Cullen, Jaya M. Mehra, Ling T. Guo, and Katie M. Minor. 2024. "Multi-Allelic Mitochondrial DNA Deletions in an Adult Dog with Chronic Weakness, Exercise Intolerance and Lactic Acidemia" Animals 14, no. 13: 1946. https://doi.org/10.3390/ani14131946
APA StyleShelton, G. D., Mickelson, J. R., Friedenberg, S. G., Cullen, J. N., Mehra, J. M., Guo, L. T., & Minor, K. M. (2024). Multi-Allelic Mitochondrial DNA Deletions in an Adult Dog with Chronic Weakness, Exercise Intolerance and Lactic Acidemia. Animals, 14(13), 1946. https://doi.org/10.3390/ani14131946