
Transcriptional Signatures of Domestication Revealed through Meta-Analysis of Pig, Chicken, Wild Boar, and Red Junglefowl Gene Expression Data


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Curation of Gene Expression Data from Public Databases
2.2. Gene Expression Quantification
2.3. Calculation of DW-Ratio
2.4. Classification of Differentially Expressed Genes (DEGs)
2.5. Calculation of DW-Scores
2.6. Gene Set Enrichment Analysis
2.7. Commonly Upregulated Genes in Domesticated Animals and Wild Ancestors
3. Results
3.1. Overview of This Study
3.2. Curation and Breakdown of RNA-Seq Data Obtained from Public Databases
3.3. Characterization of DEGs and Enrichment Analysis of Pigs and Boars
3.4. Characterization of DEGs and Enrichment Analysis in Chickens and Red Junglefowl
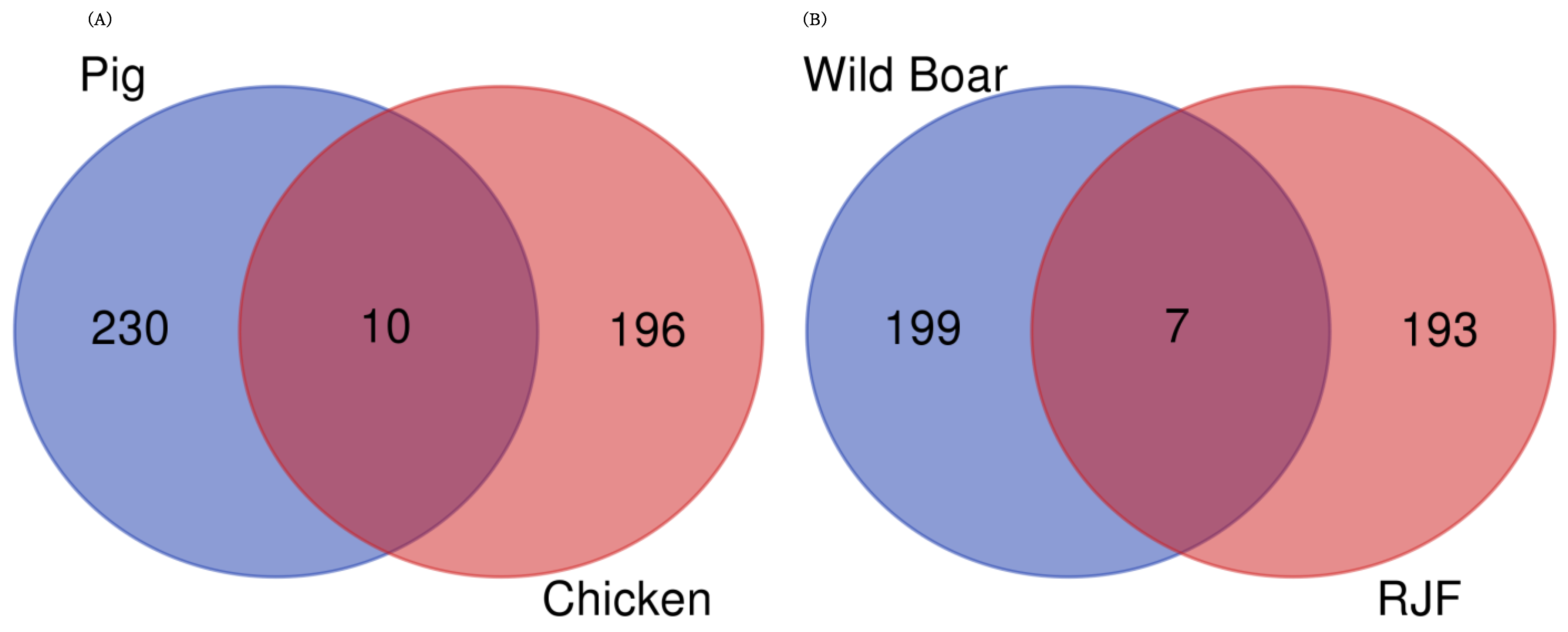
3.5. Common DEGs in Domesticated Animals and Their Wild Ancestors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Dijk, M.; Morley, T.; Rau, M.L.; Saghai, Y. A Meta-Analysis of Projected Global Food Demand and Population at Risk of Hunger for the Period 2010–2050. Nat. Food 2021, 2, 494–501. [Google Scholar] [CrossRef] [PubMed]
- OECD. OECD-FAO Agricultural Outlook 2023–2032; Organisation for Economic Co-Operation and Development: Paris, France, 2023. [Google Scholar]
- Ulmer, J.B.; Valley, U.; Rappuoli, R. Vaccine Manufacturing: Challenges and Solutions. Nat. Biotechnol. 2006, 24, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Giuffra, E.; Kijas, J.M.H.; Amarger, V.; Carlborg, Ö.; Jeon, J.-T.; Andersson, L. The Origin of the Domestic Pig: Independent Domestication and Subsequent Introgression. Genetics 2000, 154, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Lawal, R.A.; Martin, S.H.; Vanmechelen, K.; Vereijken, A.; Silva, P.; Al-Atiyat, R.M.; Aljumaah, R.S.; Mwacharo, J.M.; Wu, D.-D.; Zhang, Y.-P.; et al. The Wild Species Genome Ancestry of Domestic Chickens. BMC Biol. 2020, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Quiniou, N.; Dagorn, J.; Gaudré, D. Variation of Piglets’ Birth Weight and Consequences on Subsequent Performance. Livest. Prod. Sci. 2002, 78, 63–70. [Google Scholar] [CrossRef]
- Zuidhof, M.J.; Schneider, B.L.; Carney, V.L.; Korver, D.R.; Robinson, F.E. Growth, Efficiency, and Yield of Commercial Broilers from 1957, 1978, and 2005. Poult. Sci. 2014, 93, 2970–2982. [Google Scholar] [CrossRef] [PubMed]
- Olkowski, A.A.; Laarveld, B.; Wojnarowicz, C.; Chirino-Trejo, M.; Chapman, D.; Wysokinski, T.W.; Quaroni, L. Biochemical and Physiological Weaknesses Associated with the Pathogenesis of Femoral Bone Degeneration in Broiler Chickens. Avian Pathol. 2011, 40, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, S.; Morozumi, T.; Shimanuki, S.-I.; Hayashi, T.; Uenishi, H.; Domukai, M.; Okumura, N.; Awata, T. Fine Mapping of a Swine Quantitative Trait Locus for Number of Vertebrae and Analysis of an Orphan Nuclear Receptor, Germ Cell Nuclear Factor (NR6A1). Genome Res. 2007, 17, 586–593. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.-J. Regulation of Skeletal Muscle Mass in Mice by a New TGF-p Superfamily Member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Bono, H.; Hirota, K. Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines 2020, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—From Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]
- Download SRA Sequences from Entrez Search Results. Available online: https://www.ncbi.nlm.nih.gov/sra/docs/sradownload/ (accessed on 1 June 2023).
- Krueger, F. Babraham Bioinformatics-Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 1 July 2023).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Tximport. Available online: http://bioconductor.org/packages/tximport/ (accessed on 1 September 2023).
- Ono, Y.; Bono, H. Multi-Omic Meta-Analysis of Transcriptomes and the Bibliome Uncovers Novel Hypoxia-Inducible Genes. Biomedicines 2021, 9, 582. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Draw Venn Diagram. Available online: https://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 1 January 2024).
- Jeon, J.-T.; Carlborg, Ö.; Törnsten, A.; Giuffra, E.; Amarger, V.; Chardon, P.; Andersson-Eklund, L.; Andersson, K.; Hansson, I.; Lundström, K.; et al. A Paternally Expressed QTL Affecting Skeletal and Cardiac Muscle Mass in Pigs Maps to the IGF2 Locus. Nat. Genet. 1999, 21, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Nezer, C.; Moreau, L.; Brouwers, B.; Coppieters, W.; Detilleux, J.; Hanset, R.; Karim, L.; Kvasz, A.; Leroy, P.; Georges, M. An Imprinted QTL with Major Effect on Muscle Mass and Fat Deposition Maps to the IGF2 Locus in Pigs. Nat. Genet. 1999, 21, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, A.-S.; Nguyen, M.; Braunschweig, M.; Nezer, C.; Collette, C.; Moreau, L.; Archibald, A.L.; Haley, C.S.; Buys, N.; Tally, M.; et al. A Regulatory Mutation in IGF2 Causes a Major QTL Effect on Muscle Growth in the Pig. Nature 2003, 425, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; He, H.; Shen, X.; Zhao, J.; Cao, X.; Han, S.; Cui, C.; Chen, Y.; Wei, Y.; Xia, L.; et al. miR-9-5p Inhibits Skeletal Muscle Satellite Cell Proliferation and Differentiation by Targeting IGF2BP3 through the IGF2-PI3K/Akt Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 1655. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A Comprehensive Epigenome Atlas Reveals DNA Methylation Regulating Skeletal Muscle Development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Cuvier, O.; Hirano, T. Chromosome Condensation by a Human Condensin Complex in Xenopus Egg Extracts. J. Biol. Chem. 2001, 276, 5417–5420. [Google Scholar] [CrossRef]
- Tetens, J.; Widmann, P.; Kühn, C.; Thaller, G. A Genome-Wide Association Study Indicates LCORL/NCAPG as a Candidate Locus for Withers Height in German Warmblood Horses. Anim. Genet. 2013, 44, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Signer-Hasler, H.; Flury, C.; Haase, B.; Burger, D.; Simianer, H.; Leeb, T.; Rieder, S. A Genome-Wide Association Study Reveals Loci Influencing Height and Other Conformation Traits in Horses. PLoS ONE 2012, 7, e37282. [Google Scholar] [CrossRef] [PubMed]
- Lindholm-Perry, A.K.; Sexten, A.K.; Kuehn, L.A.; Smith, T.P.; King, D.A.; Shackelford, S.D.; Wheeler, T.L.; Ferrell, C.L.; Jenkins, T.G.; Snelling, W.M.; et al. Association, Effects and Validation of Polymorphisms within the NCAPG—LCORL Locus Located on BTA6 with Feed Intake, Gain, Meat and Carcass Traits in Beef Cattle. BMC Genet. 2011, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Hoshiba, H.; Setoguchi, K.; Watanabe, T.; Kinoshita, A.; Mizoshita, K.; Sugimoto, Y.; Takasuga, A. Comparison of the Effects Explained by Variations in the Bovine PLAG1 and NCAPG Genes on Daily Body Weight Gain, Linear Skeletal Measurements and Carcass Traits in Japanese Black Steers from a Progeny Testing Program. Anim. Sci. J. 2013, 84, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Guo, Y.; Zhang, L.; Xu, L.; Gao, X.; Zhu, B.; Gao, H.; Ni, H.; Chen, Y. Multi-Strategy Genome-Wide Association Studies Identify the DCAF16-NCAPG Region as a Susceptibility Locus for Average Daily Gain in Cattle. Sci. Rep. 2016, 6, 38073. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Duan, X.; Chen, S.; He, H.; Liu, X. NCAPG Is Differentially Expressed during Longissimus Muscle Development and Is Associated with Growth Traits in Chinese Qinchuan Beef Cattle. Genet. Mol. Biol. 2015, 38, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, K.; Watanabe, T.; Weikard, R.; Albrecht, E.; Kühn, C.; Kinoshita, A.; Sugimoto, Y.; Takasuga, A. The SNP c.1326T>G in the Non-SMC Condensin I Complex, Subunit G (NCAPG) Gene Encoding a p.Ile442Met Variant Is Associated with an Increase in Body Frame Size at Puberty in Cattle. Anim. Genet. 2011, 42, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Al-Mamun, H.A.; Kwan, P.; Clark, S.A.; Ferdosi, M.H.; Tellam, R.; Gondro, C. Genome-Wide Association Study of Body Weight in Australian Merino Sheep Reveals an Orthologous Region on OAR6 to Human and Bovine Genomic Regions Affecting Height and Weight. Genet. Sel. Evol. 2015, 47, 66. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Jeong, J.-Y.; An, Y.J.; Lee, J.-H.; Yim, H.-S. FGF11 Influences 3T3-L1 Preadipocyte Differentiation by Modulating the Expression of PPARγ Regulators. FEBS Open Bio 2019, 9, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR Gamma 2: Tissue-Specific Regulator of an Adipocyte Enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Zhou, Y.; Yang, J.; Li, J.; Peng, Y.; Zhang, X.; Miao, Y.; Jiang, W.; Bu, G.; Hou, L.; et al. Targeted Overexpression of PPARγ in Skeletal Muscle by Random Insertion and CRISPR/Cas9 Transgenic Pig Cloning Enhances Oxidative Fiber Formation and Intramuscular Fat Deposition. FASEB J. 2021, 35, e21308. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, M.; Yuan, L.; Xu, Y.; Zhou, H.; Lian, Z.; Liu, P.; Li, X. LGP2 Promotes Type I Interferon Production To Inhibit PRRSV Infection via Enhancing MDA5-Mediated Signaling. J. Virol. 2023, 97, e01843-22. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-Order Substrate Recognition of eIF2α by the RNA-Dependent Protein Kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Minks, M.A.; West, D.K.; Benvin, S.; Baglioni, C. Structural Requirements of Double-Stranded RNA for the Activation of 2′,5′-Oligo(A) Polymerase and Protein Kinase of Interferon-Treated HeLa Cells. J. Biol. Chem. 1979, 254, 10180–10183. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, R.; Wang, W.; Wu, Z.; Dai, Z.; Wang, J.; Wu, G. Biochemical and Physiological Bases for Utilization of Dietary Amino Acids by Young Pigs. J. Anim. Sci. Biotechnol. 2013, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Suryawan, A.; Jeyapalan, A.S.; Orellana, R.A.; Wilson, F.A.; Nguyen, H.V.; Davis, T.A. Leucine Stimulates Protein Synthesis in Skeletal Muscle of Neonatal Pigs by Enhancing mTORC1 Activation. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E868–E875. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, M.; Lee, J.; He, C.; Xie, Z. Leucine Supplementation Increases SIRT1 Expression and Prevents Mitochondrial Dysfunction and Metabolic Disorders in High-Fat Diet-Induced Obese Mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1234–E1244. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.-W.; Han, S.-F.; Zhang, J.-W.; Xu, J.-Y.; Qin, L.-Q. Leucine Supplementation Improves Leptin Sensitivity in High-Fat Diet Fed Rats. Food Nutr. Res. 2015, 59, 27373. [Google Scholar] [CrossRef] [PubMed]
- Kamarul, T.; Abdul Rahim, S.; Tumin, M.; Selvaratnam, L.; Ahmad, T.S. A Preliminary Study of the Effects of Glucosamine Sulphate and Chondroitin Sulphate on Surgically Treated and Untreated Focal Cartilage Damage. Eur. Cells Mater. J. 2011, 21, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.M.S.; dos Santos Neto, L.D.; Sgavioli, S.; Araújo, I.C.S.; Reis, A.A.S.; Santos, R.S.; de Araújo, E.G.; Leandro, N.S.M.; Café, M.B. Effect of Glycosaminoglycans on the Structure and Composition of Articular Cartilage and Bone of Broilers. Poult. Sci. 2023, 102, 102916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, C.X.; Chamba, Y.; Ling, Y. Blood Characteristics for High Altitude Adaptation in Tibetan Chickens1. Poult. Sci. 2007, 86, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Bokkers, E.A.M.; Koene, P. Behaviour of Fast- and Slow Growing Broilers to 12 Weeks of Age and the Physical Consequences. Appl. Anim. Behav. Sci. 2003, 81, 59–72. [Google Scholar] [CrossRef]
- Wan, L.; Wang, S.; Xie, Z.; Ren, H.; Xie, L.; Luo, S.; Li, M.; Xie, Z.; Fan, Q.; Zeng, T.; et al. Chicken IFI6 Inhibits Avian Reovirus Replication and Affects Related Innate Immune Signaling Pathways. Front. Microbiol. 2023, 14, 1237438. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; et al. Identification and Molecular Characterization of Fractalkine Receptor CX3CR1, Which Mediates Both Leukocyte Migration and Adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.J.; Hahn, C.S.; Kim, K.I.; Yan, M.; Rosario, D.; Li, L.; de la Torre, J.C.; Zhang, D.-E. Role of ISG15 Protease UBP43 (USP18) in Innate Immunity to Viral Infection. Nat. Med. 2004, 10, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Gunn, M.D.; Ngo, V.N.; Ansel, K.M.; Ekland, E.H.; Cyster, J.G.; Williams, L.T. A B-Cell-Homing Chemokine Made in Lymphoid Follicles Activates Burkitt’s Lymphoma Receptor-1. Nature 1998, 391, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Clarke, E.; Rahman, N.; Page, N.; Rolph, M.S.; Stewart, G.J.; Jones, G.J. Functional Characterization of the Atopy-Associated Gene PHF11. J. Allergy Clin. Immunol. 2008, 121, 1148–1154.e3. [Google Scholar] [CrossRef] [PubMed]
- El-Diwany, R.; Soliman, M.; Sugawara, S.; Breitwieser, F.; Skaist, A.; Coggiano, C.; Sangal, N.; Chattergoon, M.; Bailey, J.R.; Siliciano, R.F.; et al. CMPK2 and BCL-G Are Associated with Type 1 Interferon–Induced HIV Restriction in Humans. Sci. Adv. 2018, 4, eaat0843. [Google Scholar] [CrossRef] [PubMed]
- Mukilan, M.; Rajathei, D.M.; Jeyaraj, E.; Kayalvizhi, N.; Rajan, K.E. MiR-132 Regulated Olfactory Bulb Proteins Linked to Olfactory Learning in Greater Short-Nosed Fruit Bat Cynopterus Sphinx. Gene 2018, 671, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb Group Protein EZH2 Directly Controls DNA Methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Cai, Y.; Yang, Y.; Yan, X.; Liu, X.; Zhou, T. Centrosomal Protein FOR20 Is Essential for S-Phase Progression by Recruiting Plk1 to Centrosomes. Cell Res. 2013, 23, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.W.; Mayer, U.; Nischt, R.; Aumailley, M.; Reinhardt, D.; Wiedemann, H.; Mann, K.; Timpl, R.; Krieg, T.; Engel, J. Recombinant Nidogen Consists of Three Globular Domains and Mediates Binding of Laminin to Collagen Type IV. EMBO J. 1991, 10, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic Evidence That Small Maf Proteins Are Essential for the Activation of Antioxidant Response Element-Dependent Genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Seguin, S.J.; Lambert, H.; Landry, J. HspB8 Chaperone Activity toward Poly(Q)-Containing Proteins Depends on Its Association with Bag3, a Stimulator of Macroautophagy. J. Biol. Chem. 2008, 283, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Elsafadi, M.; Manikandan, M.; Dawud, R.A.; Alajez, N.M.; Hamam, R.; Alfayez, M.; Kassem, M.; Aldahmash, A.; Mahmood, A. Transgelin Is a TGFβ-Inducible Gene That Regulates Osteoblastic and Adipogenic Differentiation of Human Skeletal Stem Cells through Actin Cytoskeleston Organization. Cell Death Dis. 2016, 7, e2321. [Google Scholar] [CrossRef] [PubMed]
- Kirchmaier, B.C.; Poon, K.L.; Schwerte, T.; Huisken, J.; Winkler, C.; Jungblut, B.; Stainier, D.Y.; Brand, T. The Popeye Domain Containing 2 (Popdc2) Gene in Zebrafish Is Required for Heart and Skeletal Muscle Development. Dev. Biol. 2012, 363, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Westenbroek, R.E.; Xu, X.; Edwards, C.A.; Sorenson, D.R.; Chen, Y.; McEwen, D.P.; O’Malley, H.A.; Bharucha, V.; Meadows, L.S.; et al. Mice Lacking Sodium Channel Β1 Subunits Display Defects in Neuronal Excitability, Sodium Channel Expression, and Nodal Architecture. J. Neurosci. 2004, 24, 4030–4042. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Karlsson, H.K.R.; Szekeres, F.; Chibalin, A.V.; Krook, A.; Zierath, J.R. Suppression of 5′-Nucleotidase Enzymes Promotes AMP-Activated Protein Kinase (AMPK) Phosphorylation and Metabolism in Human and Mouse Skeletal Muscle. J. Biol. Chem. 2011, 286, 34567–34574. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | Description |
|---|---|---|
| ENSG00000106462 | EZH2 | Enhancer of zeste 2 polycomb repressive complex 2 subunit |
| ENSG00000126709 | IFI6 | Interferon alpha inducible protein 6 |
| ENSG00000184979 | USP18 | Ubiquitin specific peptidase 18 |
| ENSG00000099256 | PRTFDC1 | Phosphoribosyl transferase domain containing 1 |
| ENSG00000133393 | CEP20 | Centrosomal protein 20 |
| ENSG00000116962 | NID1 | Nidogen 1 |
| ENSG00000156234 | CXCL13 | C-X-C motif chemokine ligand 13 |
| ENSG00000134326 | CMPK2 | Cytidine/uridine monophosphate kinase 2 |
| ENSG00000168329 | CX3CR1 | C-X3-C motif chemokine receptor 1 |
| ENSG00000136147 | PHF11 | PHD finger protein 11 |
| Gene ID | Gene Name | Description |
|---|---|---|
| ENSG00000149591 | TAGLN | Transgelin |
| ENSG00000121577 | POPDC2 | Popeye domain containing 2 |
| ENSG00000185022 | MAFF | MAF bZIP transcription factor F |
| ENSG00000152137 | HSPB8 | Heat shock protein family B (small) member 8 |
| ENSG00000105711 | SCN1B | Sodium voltage-gated channel beta subunit 1 |
| ENSG00000116981 | NT5C1A | 5′-nucleotidase, cytosolic IA |
| ENSG00000151929 | BAG3 | BAG cochaperone 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uno, M.; Bono, H. Transcriptional Signatures of Domestication Revealed through Meta-Analysis of Pig, Chicken, Wild Boar, and Red Junglefowl Gene Expression Data. Animals 2024, 14, 1998. https://doi.org/10.3390/ani14131998
Uno M, Bono H. Transcriptional Signatures of Domestication Revealed through Meta-Analysis of Pig, Chicken, Wild Boar, and Red Junglefowl Gene Expression Data. Animals. 2024; 14(13):1998. https://doi.org/10.3390/ani14131998
Chicago/Turabian StyleUno, Motoki, and Hidemasa Bono. 2024. "Transcriptional Signatures of Domestication Revealed through Meta-Analysis of Pig, Chicken, Wild Boar, and Red Junglefowl Gene Expression Data" Animals 14, no. 13: 1998. https://doi.org/10.3390/ani14131998
APA StyleUno, M., & Bono, H. (2024). Transcriptional Signatures of Domestication Revealed through Meta-Analysis of Pig, Chicken, Wild Boar, and Red Junglefowl Gene Expression Data. Animals, 14(13), 1998. https://doi.org/10.3390/ani14131998