
Mitochondrial Genomes of Streptopelia decaocto: Insights into Columbidae Phylogeny
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Laboratory Analyses
2.2. Genome Sequencing and Annotation
2.3. Phylogenetic Analysis of the Cytb Gene
3. Results
3.1. Genome or Ganization: Structure and Composition
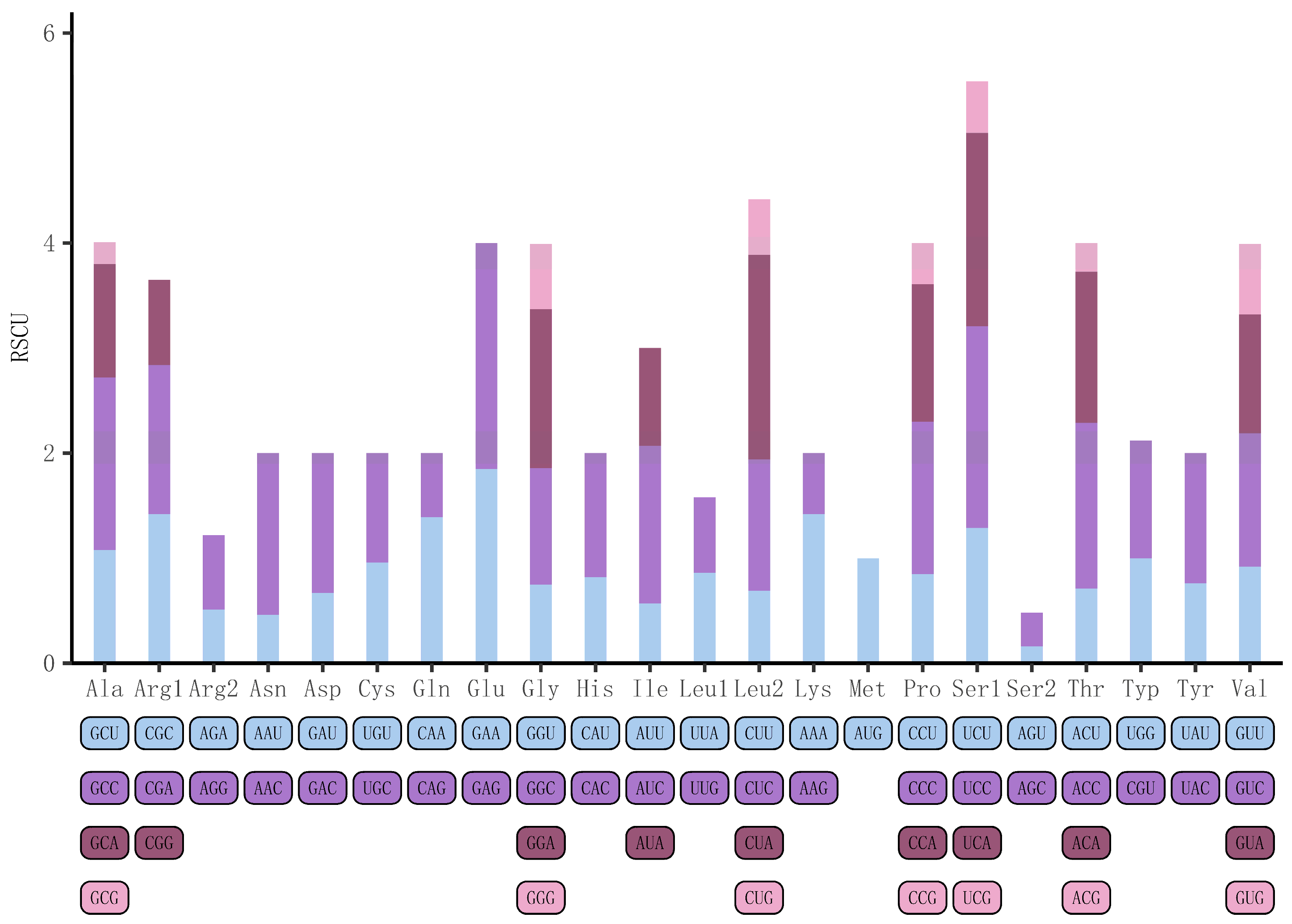
3.2. The Protein-Coding Genes
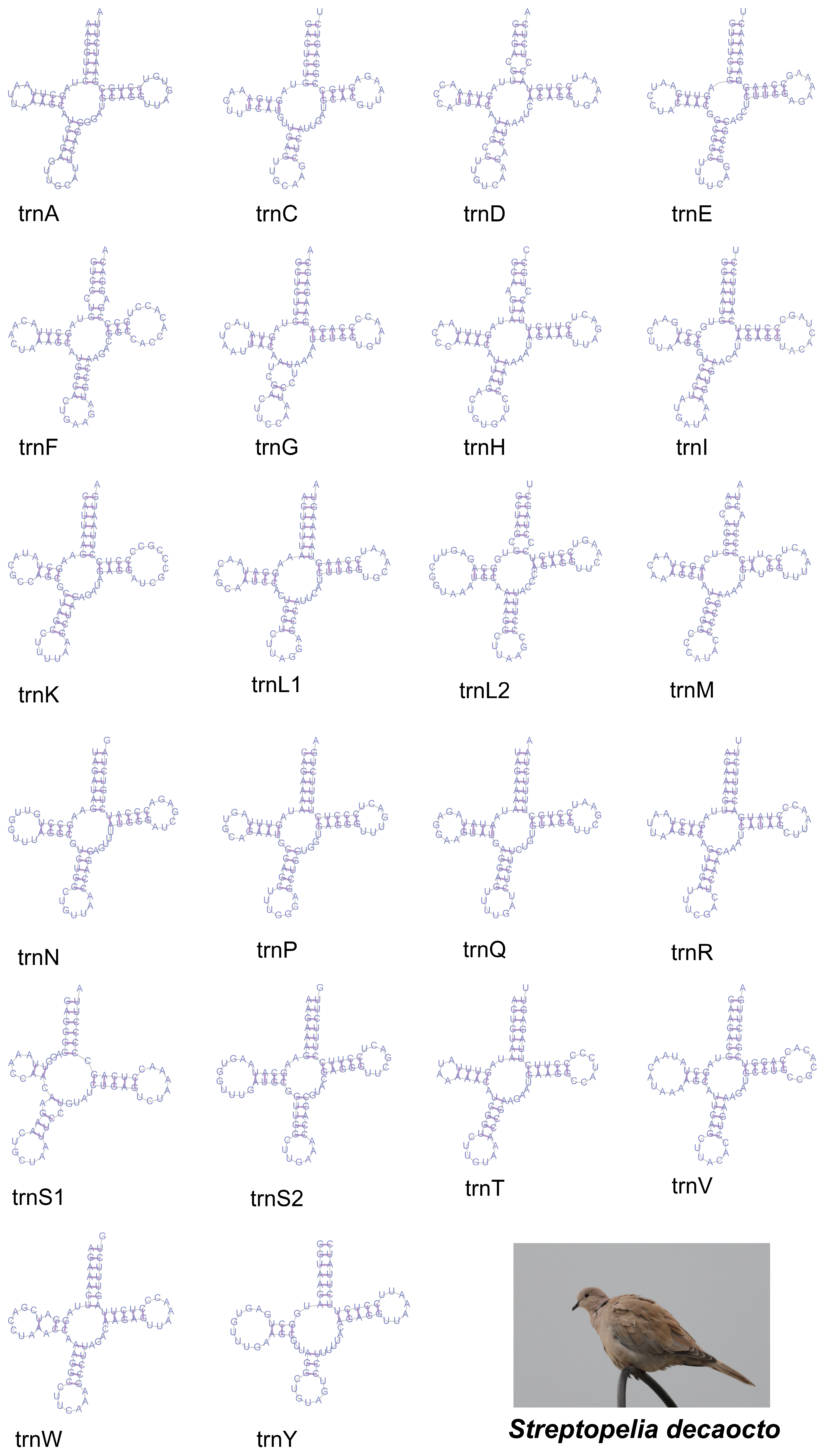
3.3. Transfer RNA and Ribosomal RNA Genes
3.4. Anticodon, Non-Coding Regions, and Control Regions
3.5. Mitogenomes of Columbidae Species
3.6. Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Soares, A.E.; Novak, B.J.; Haile, J.; Heupink, T.H.; Fjeldså, J.; Gilbert, M.T.P.; Poinar, H.; Church, G.M.; Shapiro, B. Complete mitochondrial genomes of living and extinct pigeons revise the timing of the columbiform radiation. BMC Evol. Biol. 2016, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, R.; Furo, I.d.O.; Gomes, A.J.B.; Kiazim, L.G.; Gunski, R.J.; Garnero, A.d.V.; Pereira, J.C.; Ferguson-Smith, M.A.; de Oliveira, E.H.C.; Griffin, D.K.; et al. A comprehensive cytogenetic analysis of several members of the family Columbidae (Aves, Columbiformes). Genes 2020, 11, 632. [Google Scholar] [CrossRef] [PubMed]
- Floigl, K.; Benedetti, Y.; Reif, J.; Morelli, F. Spatial Distribution and habitat overlap of five columbidae species in the Czech Republic. Animals 2022, 12, 743. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Zhang, Z.; Ding, P.; Ding, C.; Lu, X.; Zhang, Y. A Checklist on the Classification and Distribution of the Birds of the World; Science Press: Beijing, China, 2002. [Google Scholar]
- BirdLife International. Species Factsheet: Ptilinopus arcanus; BirdLife International: Oxford, UK, 2024. [Google Scholar]
- BirdLife International. Species Factsheet: Ectopistes migratorius; BirdLife International: Oxford, UK, 2024. [Google Scholar]
- BirdLife International. Species Factsheet: Alectroenas pulcherrimus; BirdLife International: Oxford, UK, 2024. [Google Scholar]
- BirdLife International. Species Factsheet: Streptopelia decaocto; BirdLife International: Oxford, UK, 2024. [Google Scholar]
- Mackinnon, J.; Showler, D.; Phillipps, K.; Mackinnon, J. A Field Guide to the Birds of China: Ornithology; Oxford University Press: Oxford, UK, 2000; Volume 18, pp. 841–843. [Google Scholar]
- Lesaffre, G. Merveilleux Oiseaux; Rustica: Paris, France, 2020. [Google Scholar]
- Song, S.; Bao, S.; Wang, Y.; Bao, X.; An, B.; Wang, X.; Liu, N. Population structure and demographic history of the chukar partridge Alectoris chukar in China. Curr. Zool. 2013, 59, 458–474. [Google Scholar] [CrossRef]
- Nagamitsu, T.; Yasuda, M.; Saito-Morooka, F.; Inoue, M.N.; Nishiyama, M.; Goka, K.; Sugiura, S.; Maeto, K.; Okabe, K.; Taki, H. Genetic structure and potential environmental determinants of local genetic diversity in Japanese Honeybees (Apis cerana japonica). PLoS ONE 2016, 11, e0167233. [Google Scholar] [CrossRef]
- Donne-Goussé, C.; Laudet, V.; Hänni, C. A molecular phylogeny of anseriformes based on mitochondrial DNA analysis. Mol. Phylogenet. Evol. 2002, 23, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y. Mitochondrial Whole Genome Sequence Analysis of 6 Species of Birds; Ludong University: Yantai, China, 2015. [Google Scholar]
- Tamashiro, R.A.; White, N.D.; Braun, M.J.; Faircloth, B.C.; Braun, E.L.; Kimball, R.T. What are the roles of taxon sampling and model fit in tests of cytb-nuclear discordance using avian mitogenomic data. Mol. Phylogenet. Evol. 2019, 130, 132–142. [Google Scholar] [CrossRef]
- Powell, A.F.; Barker, F.K.; Lanyon, S.M. Empirical evaluation of partitioning schemes for phylogenetic analyses of mitogenomic data: An avian case study. Mol. Phylogenet. Evol. 2013, 66, 69–79. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Danielson, M.J.; Faircloth, B.C.; Glenn, T.C.; Braun, E.L.; Kimball, R.T. Incongruence among different mitochondrial regions: A case study using complete mitogenomes. Mol. Phylogenet. Evol. 2014, 78, 314–323. [Google Scholar] [CrossRef]
- Urantówka, A.D.; Kroczak, A.; Mackiewicz, P. Complete mitochondrial genome of bronze-winged parrot (Pionus chalcopterus chalcopterus, Psittaciformes). Mitochondrial DNA B Resour. 2017, 2, 744–746. [Google Scholar] [CrossRef]
- Anmarkrud, J.A.; Lifjeld, J.T. Complete mitochondrial genomes of eleven extinct or possibly extinct bird species. Mol. Ecol. Resour. 2016, 17, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.H.; Shi, H.R.; Kang, S.G.; Hwang, U.W. Complete mitochondrial genome of the Japanese wood pigeon, Columba janthina janthina (Columbiformes, Columbidae). Mitochondrial DNA 2016, 27, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Kan, X.; Li, X.; Zhang, L.; Chen, L.; Qian, C.; Zhang, X.; Wang, L. Characterization of the complete mitochondrial genome of the Rock pigeon, Columba livia (Columbiformes: Columbidae). Genet. Mol. Res. 2010, 9, 1234–1249. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.A.; Battistuzzi, F.U.; Lentino, M.; Aguilar, R.F.; Kumar, S.; Escalante, A.A. Evolution of modern birds revealed by mitogenomics: Timing the radiation and origin of major orders. Mol. Biol. Evol. 2011, 28, 1927–1942. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.Q.; Guo, P.C.; Li, Y.M.; Qi, S.M.; Bai, C.Y.; Zhao, Z.H.; Sun, J.H. Complete mitochondrial genome of the Spotted dove (Streptopelia chinensis). Mitochondrial DNA Part A 2016, 27, 3067–3068. [Google Scholar] [CrossRef] [PubMed]
- Besnard, G.; Bertrand, J.A.; Delahaie, B.; Bourgeois, Y.X.; Lhuillier, E.; Thébaud, C. Valuing museum specimens: High-throughput DNA sequencing on historical collections of New Guinea crowned pigeons (Goura). Biol. J. Linn. Soc. 2016, 117, 71–82. [Google Scholar] [CrossRef]
- Chen, Y.X.; Sun, C.H.; Li, Y.K.; Fei, Y.L.; Xue, X.M.; Hou, S.L.; Zhou, Y.W.; Jiang, J.; Guo, H.T. Complete mitogenome of Treron sphenurus (Aves, Columbiformes): The first representative from the genus Treron, genomic comparisons and phylogenetic analysis of Columbidae. Anim. Biotechnol. 2022, 33, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Calderón, L.; Campagna, L.; Wilke, T.; Lormee, H.; Eraud, C.; Dunn, J.C.; Rocha, G.; Zehtindjiev, P.; Bakaloudis, D.E.; Metzger, B.; et al. Genomic evidence of demographic fluctuations and lack of genetic structure across flyways in a long distance migrant, the European turtle dove. BMC Evol. Biol. 2016, 16, 237. [Google Scholar] [CrossRef] [PubMed]
- Banks, R.C.; Weckstein, J.D.; Remsen, J.V., Jr.; Johnson, K.P. Classification of a clade of New World doves (Columbidae: Zenaidini). Zootaxa 2013, 3669, 184–188. [Google Scholar] [CrossRef]
- Huang, Z.; Tu, F.; Liu, X. Determination of the complete mitogenome of spotted dove, Spilopelia chinensis (Columbiformes: Columbidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 4224–4225. [Google Scholar] [CrossRef]
- Sorensen, M.; Sanz, A.; Gómez, J.; Pamplona, R.; Barja, G. Effects of fasting on oxidative stress in rat liver mitochondria. Free Radic. Res. 2006, 40, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Borgström, E.; Lundin, S.; Lundeberg, J. Large scale library generation for high throughput sequencing. PLoS ONE 2011, 6, e19119. [Google Scholar] [CrossRef] [PubMed]
- Cronn, R.; Liston, A.; Parks, M.; Gernandt, D.S.; Shen, R.; Mockler, T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008, 36, e122. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Nandi, T.; Ong, C.; Singh, A.P.; Boddey, J.; Atkins, T.; Sarkar-Tyson, M.; Essex-Lopresti, A.E.; Chua, H.H.; Pearson, T.; Kreisberg, J.F.; et al. A genomic survey of positive selection in Burkholderia pseudomallei provides insights into the evolution of accidental virulence. PLoS Pathog. 2010, 6, e1000845. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Nylander, J. MrModeltest V2. Program Distributed by the Author. Bioinformatics 2004, 24, 581–583. Available online: https://www.researchgate.net/publication/285805344_MrModeltest_V2_Program_Distributed_by_the_Author (accessed on 25 July 2024). [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.H.; Tu, F.Y.; Murphy, R.W. Analysis of the complete mitogenome of Oriental turtle dove (Streptopelia orientalis) and implications for species divergence. Biochem. Syst. Ecol. 2016, 65, 209–213. [Google Scholar] [CrossRef]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.H. Mutation and selection on the anticodon of tRNA genes in vertebrate mitochondrial genomes. Gene 2005, 345, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Bruxaux, J.; Gabrielli, M.; Ashari, H.; Prŷs-Jones, R.; Joseph, L.; Milá, B.; Besnard, G.; Thébaud, C. Recovering the evolutionary history of crowned pigeons (Columbidae: Goura): Implications for the biogeography and conservation of New Guinean lowland birds. Mol. Phylogenet. Evol. 2018, 120, 248–258. [Google Scholar] [CrossRef]
- Feng, S.; Fang, Q.; Barnett, R.; Li, C.; Han, S.; Kuhlwilm, M.; Zhou, L.; Pan, H.; Deng, Y.; Chen, G.; et al. The Genomic Footprints of the Fall and Recovery of the Crested Ibis. Curr. Biol. 2019, 29, 340–349.e347. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Ebert, D. Covariation of mitochondrial genome size with gene lengths: Evidence for gene length reduction during mitochondrial evolution. J. Mol. Evol. 2004, 59, 90–96. [Google Scholar] [CrossRef]
- Selosse, M.A.; Albert, B.; Godelle, B. Reducing the genome size of organelles favours gene transfer to the nucleus. Trends Ecol. Evol. 2001, 16, 135–141. [Google Scholar] [CrossRef]
- Liu, H.Y.; Sun, C.H.; Zhu, Y.; Zhang, Q.Z. Complete mitogenomic and phylogenetic characteristics of the speckled wood-pigeon (Columba hodgsonii). Mol. Biol. Rep. 2020, 47, 3567–3576. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.A.; Arif, I.A. COI barcodes and phylogeny of doves (Columbidae family). Mitochondrial DNA 2013, 24, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, J.; Lu, X.; Teng, X.; Xing, Z.; Wang, S.; Feng, C.; Wang, X.; Wang, L. Mitochondrial Genomes of Streptopelia decaocto: Insights into Columbidae Phylogeny. Animals 2024, 14, 2220. https://doi.org/10.3390/ani14152220
Qu J, Lu X, Teng X, Xing Z, Wang S, Feng C, Wang X, Wang L. Mitochondrial Genomes of Streptopelia decaocto: Insights into Columbidae Phylogeny. Animals. 2024; 14(15):2220. https://doi.org/10.3390/ani14152220
Chicago/Turabian StyleQu, Jiangyong, Xiaofei Lu, Xindong Teng, Zhikai Xing, Shuang Wang, Chunyu Feng, Xumin Wang, and Lijun Wang. 2024. "Mitochondrial Genomes of Streptopelia decaocto: Insights into Columbidae Phylogeny" Animals 14, no. 15: 2220. https://doi.org/10.3390/ani14152220