
Metagenomics-Metabolomics Exploration of Three-Way-Crossbreeding Effects on Rumen to Provide Basis for Crossbreeding Improvement of Sheep Microbiome and Metabolome of Sheep
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Sample Collection
2.3. Metagenome Sequencing and Bioinformatics Analysis
2.4. Metabolome Sequencing and Bioinformatics Analysis
2.5. Data Statistics and Analysis
3. Results
3.1. Genome Profiling of Rumen Microorganisms
3.1.1. Sequencing and Diversity Analysis of Rumen Microbiota
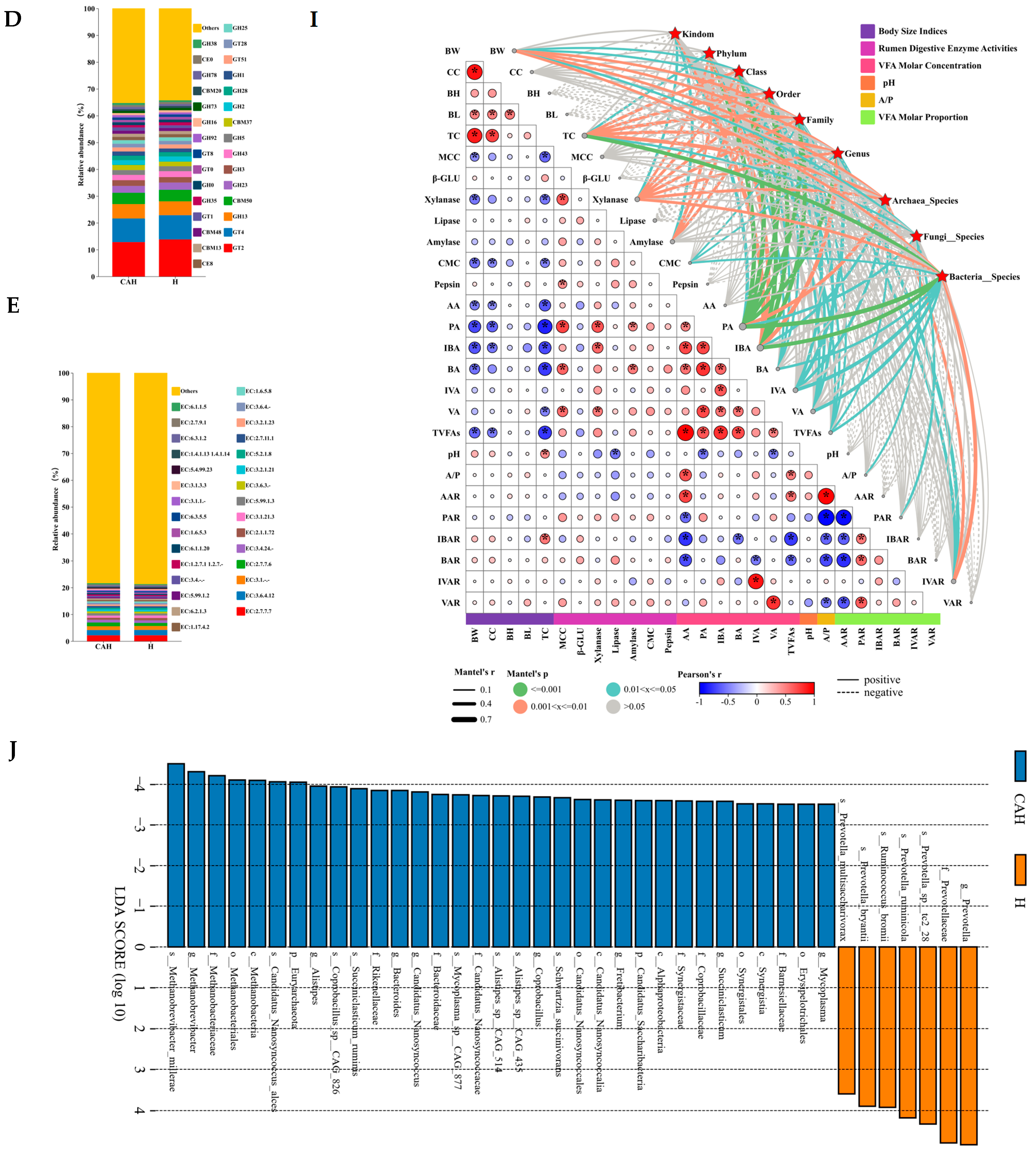
3.1.2. Analysis of Rumen Microbial Composition, Function, and Correlation
3.2. Comparison of Ruminal Metabolites in Hu and CAH Lambs
3.3. Relationship between Rumen Biomarkers and Differential Metabolites in Sheep
4. Discussion
4.1. Effect of Three-Way Crosses on the Rumen Macrogenome of Sheep
4.1.1. Effect of Three-Way Crossbreeding on Rumen Microbial Diversity in Sheep
4.1.2. Effect of Three-Way Crossbreeding on Rumen Microbial Composition in Sheep
4.1.3. Effect of Three-Way Crossbreeding on Rumen Microbial Functions in Sheep
4.2. Effect of Three-Way Crossbreeding on Rumen Metabolism in Sheep
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Yang, J.; Shen, M.; Xie, X.L.; Liu, G.J.; Xu, Y.X.; Lv, F.H.; Yang, H.; Yang, Y.L.; Liu, C.B.; et al. Whole–genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef] [PubMed]
- Akinmoladun, O.F.; Muchenje, V.; Fon, F.N.; Mpendulo, C.T. Small Ruminants: Farmers’ hope in a world threatened by water scarcity. Animals 2019, 9, 456. [Google Scholar] [CrossRef]
- Kalds, P.; Zhou, S.; Cai, B.; Liu, J.; Wang, Y.; Petersen, B.; Sonstegard, T.; Wang, X.; Chen, Y. Sheep and goat genome engineering: From random transgenesis to the CRISPR Era. Front. Genet. 2019, 10, 750. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, P.; Li, M.; Fang, W.; Yue, X.; Nanaei, H.A.; Gan, S.; Du, D.; Cai, Y.; Dai, X.; et al. A Hu sheep genome with the first ovine Y chromosome reveal introgression history after sheep domestication. Sci. China Life Sci. 2021, 64, 1116–1130. [Google Scholar] [CrossRef]
- Abegaz, S.; Bernardus, J.; Wyk, V.; Olivier, J.J. Estimation of genetic and phenotypic parameters of growth curve and their relationship with early growth and productivity in Horro sheep. Arch. Anim. Breed. 2010, 53, 85–94. [Google Scholar] [CrossRef]
- Kong, L.; Yue, Y.; Li, J.; Yang, B.; Chen, B.; Liu, J.; Lu, Z. Transcriptomics and metabolomics reveal improved performance of Hu sheep on hybridization with Southdown sheep. Food Res. Int. 2023, 173, 113240. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, D.L.; Fogarty, N.M.; Mortimer, S.I. Genetic related effects on sheep meat quality. Small Rumin. Res. 2011, 101, 160–172. [Google Scholar] [CrossRef]
- Wang, H.; Zhan, J.; Jia, H.; Jiang, H.; Pan, Y.; Zhong, X.; Zhao, S.; Huo, J. Relationship between rumen microbial differences and phenotype traits among Hu sheep and crossbred offspring Sheep. Animals 2024, 14, 1509. [Google Scholar] [CrossRef]
- de Sousa, M.A.P.; Lima, A.C.S.; Araújo, J.C.; Guimarães, C.M.C.; Joele, M.R.S.P.; Borges, I.; Daher, L.C.C.; Silva, A.G.M.E. Tissue composition and allometric growth of carcass of lambs Santa Inês and crossbreed with breed Dorper. Trop. Anim. Health Prod. 2019, 51, 1903–1908. [Google Scholar] [CrossRef]
- Lunesu, M.F.; Battacone, G.; Mellino, M.R.; Carta, S.; Pulina, G.; Nudda, A. The heavy suckling lamb of Sarda dairy sheep and its crossbreed with Dorper rams: Performance, meat quality and consumer perceptions. Meat Sci. 2023, 204, 109234. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Zhang, X.; Li, C.; Yuan, L.; Zhang, D.; Zhao, Y.; Li, X.; Cheng, J.; Lin, C.; et al. Heritability and recursive influence of host genetics on the rumen microbiota drive body weight variance in male Hu sheep lambs. Microbiome 2023, 11, 197. [Google Scholar] [CrossRef]
- Matthews, C.; Crispie, F.; Lewis, E.; Reid, M.; O’Toole, P.W.; Cotter, P.D. The rumen microbiome: A crucial consideration when optimising milk and meat production and nitrogen utilisation efficiency. Gut Microbes 2019, 10, 115–132. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Weimer, P.J. Symposium review: Host-rumen microbe interactions may be leveraged to improve the productivity of dairy cows. J. Dairy Sci. 2018, 101, 7680–7689. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Gordon, G.L.; Phillips, M.W. The role of anaerobic gut fungi in ruminants. Nutr. Res. Rev. 1998, 11, 133–168. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Li, F.; Li, C.; Zhang, D.; Li, X.; Zhao, Y.; Wang, W. Exploring the ruminal microbial community associated with fat deposition in lambs. Animals 2021, 11, 3584. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Li, F.; Li, C.; Li, G.; Zhang, D.; Song, Q.; Li, X.; Zhao, Y.; Wang, W. Characterization of the rumen microbiota and its relationship with residual feed intake in sheep. Animal 2021, 15, 100161. [Google Scholar] [CrossRef]
- Huang, Y.; Lv, H.; Song, Y.; Sun, C.; Zhang, Z.; Chen, S. Community composition of cecal microbiota in commercial yellow broilers with high and low feed efficiencies. Poult. Sci. 2021, 100, 100996. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Wang, X.; Li, F.; Zhang, D.; Li, X.; Zhao, Y.; Zhao, L.; Xu, D.; Cheng, J.; et al. Association between rumen microbiota and marbling grade in Hu sheep. Front. Microbiol. 2022, 13, 978263. [Google Scholar] [CrossRef]
- Shtossel, O.; Koren, O.; Shai, I.; Rinott, E.; Louzoun, Y. Gut microbiome-metabolome interactions predict host condition. Microbiome 2024, 12, 24. [Google Scholar] [CrossRef]
- Jansma, J.; El Aidy, S. Understanding the host-microbe interactions using metabolic modeling. Microbiome 2021, 9, 16. [Google Scholar] [CrossRef]
- Chen, L.; Zhernakova, D.V.; Kurilshikov, A.; Andreu-Sánchez, S.; Wang, D.; Augustijn, H.E.; Vich Vila, A.; Weersma, R.K.; Medema, M.H.; Netea, M.G.; et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat. Med. 2022, 28, 2333–2343. [Google Scholar] [CrossRef]
- Bar, N.; Korem, T.; Weissbrod, O.; Zeevi, D.; Rothschild, D.; Leviatan, S.; Kosower, N.; Lotan-Pompan, M.; Weinberger, A.; Le Roy, C.I.; et al. A reference map of potential determinants for the human serum metabolome. Nature 2020, 588, 135–140. [Google Scholar] [CrossRef]
- Szeligowska, N.; Cholewińska, P.; Czyż, K.; Wojnarowski, K.; Janczak, M. Inter and intraspecies comparison of the level of selected bacterial phyla in in cattle and sheep based on feces. BMC Vet. Res. 2021, 17, 224. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, X.; Fei, W.; Ye, Y.; Zhao, M.; Zheng, C. The role of short-chain fatty acids in immunity, inflammation and metabolism. Crit. Rev. Food Sci. Nutr. 2022, 62, 1–12. [Google Scholar] [CrossRef]
- Chen, Z.J. Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 2013, 14, 471–482. [Google Scholar] [CrossRef]
- Qiu, X.; Qin, X.; Chen, L.; Chen, Z.; Hao, R.; Zhang, S.; Yang, S.; Wang, L.; Cui, Y.; Li, Y.; et al. Serum biochemical parameters, rumen fermentation, and rumen bacterial communities are partly driven by the breed and sex of cattle when fed high-grain diet. Microorganisms 2022, 10, 323. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, X.; Li, F.; Liu, T.; Hu, Z.; Gao, N.; Yuan, L.; Li, X.; Zhao, Y.; Zhao, L.; et al. Whole-genome resequencing identified candidate genes associated with the number of ribs in Hu sheep. Genomics 2021, 113, 2077–2084. [Google Scholar] [CrossRef]
- Wan, Z.; Yang, H.; Cai, Y.; Ma, J.; Cheng, P.; Wang, Z.; Wang, F.; Zhang, Y. Comparative transcriptomic analysis of Hu sheep pituitary gland prolificacy at the follicular and luteal phases. Genes 2022, 13, 440. [Google Scholar] [CrossRef]
- Wang, H.B.; Zhan, J.S.; Gu, Z.Y.; Chen, X.F.; Pan, Y.; Jia, H.B.; Zhong, X.J.; Li, K.R.; Zhao, S.G.; Huo, J.H. Comparative study on meat quality characteristics of three-way hybrid sheep Charolais×Duper×Hu and Charolais×Australian White×Hu and Hu sheep. Acta Vet. Zootech. Sin. 2024, 55, 110–119. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.A.; Chang, Y.J.; Chen, S.H.; Lin, C.Y.; Ho, J.M. SQUAT: A Sequencing Quality Assessment Tool for data quality assessments of genome assemblies. BMC Genom. 2019, 19, 238. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; LI, J.Q.; WU, S.F.; ZHU, Y.P.; CHEN, Y.W.; HE, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Zhang, T.; Shen, X.T.; Liu, J.; Zhao, D.L.; Sun, Y.W.; Wang, L.; Liu, Y.J.; Gong, X.Y.; Liu, Y.X. Serum metabolomics for early diagnosis of esophageal squamous cell carcinoma by UHPLC-QTOF/MS. Metabolomics 2016, 12, 116. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Osadchiy, V.; Martin, C.R.; Mayer, E.A. The Gut-brain axis and the microbiome: Mechanisms and clinical implications. Clin. Gastroenterol. Hepatol. 2019, 17, 322–332. [Google Scholar] [CrossRef]
- Li, Z.; Wright, A.G.; Si, H.; Wang, X.; Qian, W.; Zhang, Z.; Li, G. Changes in the rumen microbiome and metabolites reveal the effect of host genetics on hybrid crosses. Environ. Microbiol. Rep. 2016, 8, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Furman, O.; Shenhav, L.; Sasson, G.; Kokou, F.; Honig, H.; Jacoby, S.; Hertz, T.; Cordero, O.X.; Halperin, E.; Mizrahi, I. Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat. Commun. 2020, 11, 1904. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, Y.; Yu, Z.; Xu, Q.; Zheng, N.; Zhao, S.; Huang, G.; Wang, J. Ruminal microbiota-host interaction and its effect on nutrient metabolism. Anim. Nutr. 2021, 7, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sha, Y.; Lv, W.; Pu, X.; Liu, X.; Luo, Y.; Hu, J.; Wang, J.; Li, S.; Zhao, Z. Sex differences in rumen fermentation and microbiota of Tibetan goat. Microb. Cell Fact. 2022, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Paul, H.A.; Bomhof, M.R.; Vogel, H.J.; Reimer, R.A. Diet-induced changes in maternal gut microbiota and metabolomic profiles influence programming of offspring obesity risk in rats. Sci. Rep. 2016, 6, 20683. [Google Scholar] [CrossRef]
- Argaw-Denboba, A.; Schmidt, T.S.B.; Di Giacomo, M.; Ranjan, B.; Devendran, S.; Mastrorilli, E.; Lloyd, C.T.; Pugliese, D.; Paribeni, V.; Dabin, J.; et al. Paternal microbiome perturbations impact offspring fitness. Nature 2024, 629, 652–659. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Kreisinger, J.; Cížková, D.; Vohánka, J.; Piálek, J. Gastrointestinal microbiota of wild and inbred individuals of two house mouse subspecies assessed using high-throughput parallel pyrosequencing. Mol. Ecol. 2014, 23, 5048–5060. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A. What is microbial community ecology? ISME J. 2009, 3, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Lei, Y.; Wu, J.; Li, Z.; Zhang, X.; Jia, L.; Wang, Y.; Ma, Y.; Zhang, K.; Cheng, Q.; et al. Antimicrobial peptides act on the rumen microbiome and metabolome affecting the performance of castrated bulls. J. Anim. Sci. Biotechnol. 2023, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhang, X.; Xu, D.; Zhang, D.; Zhang, Y.; Song, Q.; Li, X.; Zhao, Y.; Zhao, L.; Li, W.; et al. Relationship between rumen microbial differences and traits among Hu sheep, Tan sheep, and Dorper sheep. J. Anim. Sci. 2022, 100, skac261. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cheng, B.B.; Liu, Y.F.; Li, M.M.; Zhao, G.Y. Effects of red cabbage extract rich in anthocyanins on rumen fermentation, rumen bacterial community, nutrient digestion, and plasma indices in beef bulls. Animal 2022, 16, 100510. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Qi, W.; Zhang, T.; Zhang, J.; Mao, S. Multi-omics analysis revealed coordinated responses of rumen microbiome and epithelium to high-grain-induced subacute rumen acidosis in lactating dairy cows. mSystems 2022, 7, e0149021. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Wanapat, M.; Hou, F. Rumen bacteria influence milk protein yield of yak grazing on the Qinghai-Tibet plateau. Anim. Biosci. 2021, 34, 1466–1478. [Google Scholar] [CrossRef] [PubMed]
- Gharechahi, J.; Vahidi, M.F.; Bahram, M.; Han, J.L.; Ding, X.Z.; Salekdeh, G.H. Metagenomic analysis reveals a dynamic microbiome with diversified adaptive functions to utilize high lignocellulosic forages in the cattle rumen. ISME J. 2021, 15, 1108–1120. [Google Scholar] [CrossRef]
- Davis, C.K.; Webb, R.I.; Sly, L.I.; Denman, S.E.; McSweeney, C.S. Isolation and survey of novel fluoroacetate-degrading bacteria belonging to the phylum Synergistetes. FEMS Microbiol. Ecol. 2012, 80, 671–684. [Google Scholar] [CrossRef]
- Leong, L.E.; Denman, S.E.; Hugenholtz, P.; McSweeney, C.S. Amino acid and peptide utilization profiles of the fluoroacetate-degrading bacterium synergistetes strain MFA1 under varying conditions. Microb. Ecol. 2016, 71, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, A. Euryarchaeota. Encycl. Astrobiol. 2015, 764–765. [Google Scholar] [CrossRef]
- Zhang, Y.; Bobe, G.; Revel, J.S.; Rodrigues, R.R.; Sharpton, T.J.; Fantacone, M.L.; Raslan, K.; Miranda, C.L.; Lowry, M.B.; Blakemore, P.R.; et al. Improvements in metabolic syndrome by xanthohumol derivatives are linked to altered gut microbiota and bile acid metabolism. Mol. Nutr. Food Res. 2020, 64, e1900789. [Google Scholar] [CrossRef]
- Yuan, X.; Chen, R.; McCormick, K.L.; Zhang, Y.; Lin, X.; Yang, X. The role of the gut microbiota on the metabolic status of obese children. Microb. Cell Fact. 2021, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Vogel, H.; Jonas, W.; Woting, A.; Blaut, M.; Schürmann, A.; Cedernaes, J. Gut microbiota and glucometabolic alterations in response to recurrent partial sleep deprivation in normal-weight young individuals. Mol. Metab. 2016, 5, 1175–1186. [Google Scholar] [CrossRef]
- He, H.; Lin, M.; You, L.; Chen, T.; Liang, Z.; Li, D.; Xie, C.; Xiao, G.; Ye, P.; Kong, Y.; et al. Gut microbiota profile in adult patients with idiopathic nephrotic syndrome. Biomed. Res. Int. 2021, 2021, 8854969. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, N.D.; Ijaz, U.Z.; Evans, T.J. IL-17 signalling restructures the nasal microbiome and drives dynamic changes following Streptococcus pneumoniae colonization. BMC Genom. 2017, 18, 807. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, B.; Liu, Z.; Yin, Y.; Xu, G.; Han, M.; Xie, L. Effect of dietary histamine on intestinal morphology, inflammatory status, and gut microbiota in yellow catfish (Pelteobagrus fulvidraco). Fish. Shellfish. Immunol. 2021, 117, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, C.; Granata, I.; D’Argenio, V.; Tramontano, S.; Compare, D.; Guarracino, M.R.; Nardone, G.; Pilone, V.; Sacchetti, L. Characterization of the duodenal mucosal microbiome in obese adult subjects by 16S rRNA sequencing. Microorganisms 2020, 8, 485. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef]
- Lou, J.; Jiang, Y.; Rao, B.; Li, A.; Ding, S.; Yan, H.; Zhou, H.; Liu, Z.; Shi, Q.; Cui, G.; et al. Fecal microbiomes distinguish patients with autoimmune hepatitis from healthy individuals. Front. Cell Infect. Microbiol. 2020, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Gao, Y.; Wang, Y.; Liu, Q.; Sun, Z.; Fu, B.; Wen, X.; Cui, Z.; Wang, W. Diversity of a mesophilic lignocellulolytic microbial consortium which is useful for enhancement of biogas production. Bioresour. Technol. 2012, 111, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.H.; Zhang, R.K.; Liu, R.; Li, R.G.; Sun, C.M. Cultivation and functional characterization of a deep-sea lentisphaerae representative reveals its unique physiology and ecology. Front. Mar. Sci. 2022, 9, 2296–7745. [Google Scholar] [CrossRef]
- Betancur-Murillo, C.L.; Aguilar-Marín, S.B.; Jovel, J. Prevotella: A key player in ruminal metabolism. Microorganisms 2022, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, L.; Tang, G.; Yu, J.; Chen, J.; Li, Z.; Cao, Y.; Lei, X.; Deng, L.; Wu, S.; et al. Multi-omics revealed the long-term effect of ruminal keystone bacteria and the microbial metabolome on lactation performance in adult dairy goats. Microbiome 2023, 11, 215. [Google Scholar] [CrossRef]
- Russell, J.B.; Rychlik, J.L. Factors that alter rumen microbial ecology. Science 2001, 292, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef] [PubMed]
- Indugu, N.; Vecchiarelli, B.; Baker, L.D.; Ferguson, J.D.; Vanamala, J.K.P.; Pitta, D.W. Comparison of rumen bacterial communities in dairy herds of different production. BMC Microbiol. 2017, 17, 190. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.Y.; Sun, H.Z.; Wu, X.H.; Liu, J.X.; Guan, L.L. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 2020, 8, 64. [Google Scholar] [CrossRef]
- Bauman, D.E.; Harvatine, K.J.; Lock, A.L. Nutrigenomics, rumen-derived bioactive fatty acids, and the regulation of milk fat synthesis. Annu. Rev. Nutr. 2011, 31, 299–319. [Google Scholar] [CrossRef]
- Fukami, T. Historical contingency in community assembly: Integrating niches, species pools, and priority effects. Annu. Rev. Ecol. Evol. Syst. 2015, 46, 1–23. [Google Scholar] [CrossRef]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Hao, Z.; Shi, H.; Li, T.; Wang, H.; Li, Q.; Zhang, Y.; Ma, Y. Metagenomic analysis revealed differences in composition and function between liquid-associated and solid-associated microorganisms of sheep rumen. Front. Microbiol. 2022, 13, 851567. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zheng, L.; Chen, X.; Han, X.; Cao, Y.; Yao, J. Metagenomic analyses of microbial and carbohydrate-active enzymes in the rumen of dairy goats fed different rumen degradable starch. Front. Microbiol. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Xue, Y.; Lin, L.; Hu, F.; Zhu, W.; Mao, S. Disruption of ruminal homeostasis by malnutrition involved in systemic ruminal microbiota-host interactions in a pregnant sheep model. Microbiome 2020, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Slavin, J.L. Dietary fiber and body weight. Nutrition 2005, 21, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Sleeth, M.L.; Thompson, E.L.; Ford, H.E.; Zac-Varghese, S.E.; Frost, G. Free fatty acid receptor 2 and nutrient sensing: A proposed role for fibre, fermentable carbohydrates and short-chain fatty acids in appetite regulation. Nutr. Res. Rev. 2010, 23, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; An, X.; Yang, Y.; Zhang, L.; Jiao, T.; Zhao, S. Comprehensive analysis of the longissimus dorsi transcriptome and metabolome reveals the regulatory mechanism of different varieties of meat quality. J. Agric. Food Chem. 2023, 71, 1234–1245. [Google Scholar] [CrossRef]
- Wei, Y.H.; Ma, X.; Zhao, J.C.; Wang, X.Q.; Gao, C.Q. Succinate metabolism and its regulation of host-microbe interactions. Gut Microbes 2023, 15, 2190300. [Google Scholar] [CrossRef]
- Yuan, Y.; Xu, Y.; Xu, J.; Liang, B.; Cai, X.; Zhu, C.; Wang, L.; Wang, S.; Zhu, X.; Gao, P.; et al. Succinate promotes skeletal muscle protein synthesis via Erk1/2 signaling pathway. Mol. Med. Rep. 2017, 16, 7361–7366. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Zhang, X.; Chen, Q.; Xia, L. Role of succinic acid in the regulation of sepsis. Int. Immunopharmacol. 2022, 110, 109065. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zhan, J.; Jiang, H.; Jia, H.; Pan, Y.; Zhong, X.; Huo, J.; Zhao, S. Metagenomics-Metabolomics Exploration of Three-Way-Crossbreeding Effects on Rumen to Provide Basis for Crossbreeding Improvement of Sheep Microbiome and Metabolome of Sheep. Animals 2024, 14, 2256. https://doi.org/10.3390/ani14152256
Wang H, Zhan J, Jiang H, Jia H, Pan Y, Zhong X, Huo J, Zhao S. Metagenomics-Metabolomics Exploration of Three-Way-Crossbreeding Effects on Rumen to Provide Basis for Crossbreeding Improvement of Sheep Microbiome and Metabolome of Sheep. Animals. 2024; 14(15):2256. https://doi.org/10.3390/ani14152256
Chicago/Turabian StyleWang, Haibo, Jinshun Zhan, Haoyun Jiang, Haobin Jia, Yue Pan, Xiaojun Zhong, Junhong Huo, and Shengguo Zhao. 2024. "Metagenomics-Metabolomics Exploration of Three-Way-Crossbreeding Effects on Rumen to Provide Basis for Crossbreeding Improvement of Sheep Microbiome and Metabolome of Sheep" Animals 14, no. 15: 2256. https://doi.org/10.3390/ani14152256