
Moderate Genetic Diversity of MHC Genes in an Isolated Small Population of Black-and-White Snub-Nosed Monkeys (Rhinopithecus bieti)
 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Study Site and Sampling
2.2. DNA Extraction
2.3. Microsatellite Genotyping
2.4. MHC Genotyping
2.5. Data Analysis
2.5.1. Genetic Diversity
2.5.2. Selective Pressure Analysis
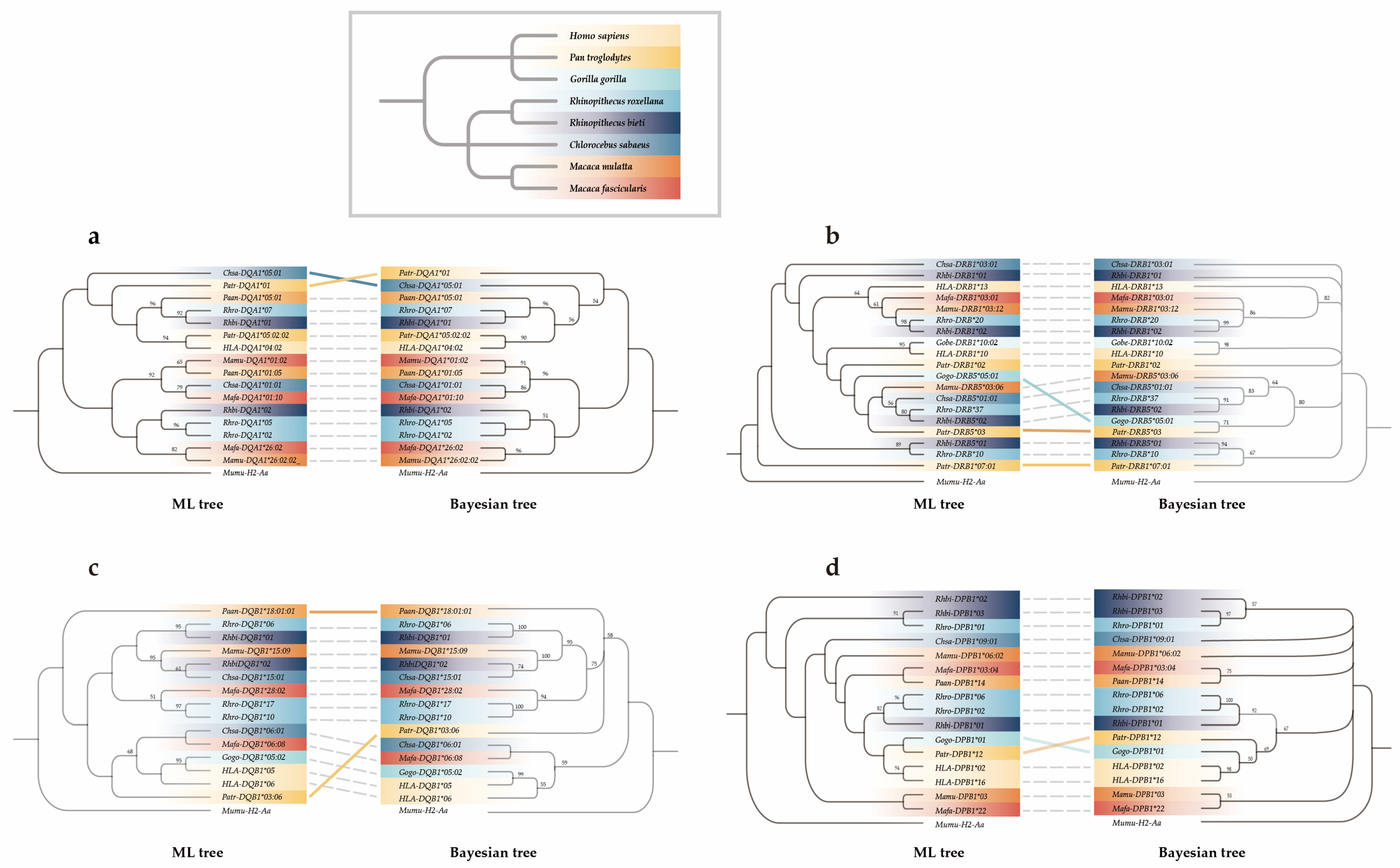
2.5.3. Phylogenetic Analysis
3. Results
3.1. MHC Allele Assignment
3.2. Genetic Variation at Microsatellites and MHC Genes
3.3. Positive Selection
3.4. Trans-Species Evolution
4. Discussion
4.1. Genetic Diversity
4.2. Historical Balancing Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bouzat, J.L. Conservation genetics of population bottlenecks: The role of chance, selection, and history. Conserv. Genet. 2010, 11, 463–478. [Google Scholar] [CrossRef]
- Pröhl, H.; Rodriguez, A. Importance of genetic-fitness correlations for the conservation of amphibians. Animals 2023, 13, 3564. [Google Scholar] [CrossRef] [PubMed]
- Blomqvist, D.; Pauliny, A.; Larsson, M.; Flodin, L.Å. Trapped in the extinction vortex? Strong genetic effects in a declining vertebrate population. BMC Evol. Biol. 2010, 10, 33. [Google Scholar] [CrossRef]
- Okamiya, H.; Kusano, T. Lower genetic diversity and hatchability in amphibian populations isolated by urbanization. Popul. Ecol. 2018, 60, 347–360. [Google Scholar] [CrossRef]
- Pearman, P.B.; Garner, T.W.J. Susceptibility of Italian agile frog populations to an emerging strain of Ranavirus parallels population genetic diversity. Ecol. Lett. 2005, 8, 401–408. [Google Scholar] [CrossRef]
- Phillips, S. Differing mortality rates in two concurrently radio-tracked populations of koala (Phascolarctos cinereus). Aust. Mammal. 2018, 40, 198–203. [Google Scholar] [CrossRef]
- Wollebaek, J.; Røed, K.H.; Brabrand, Å.; Heggenes, J. Interbreeding of genetically distinct native brown trout (Salmo trutta) populations designates offspring fitness. Aquaculture 2012, 356, 158–168. [Google Scholar] [CrossRef]
- Frankham, R. Quantitative genetics in conservation biology. Genet. Res. 1999, 74, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Gilpin, M.E.; Soule, M.E. Minimum viable populations: Processes of species extinction. In Conservation Biology: The Science of Scarcity and Diversity; Sinauer: Sunderland, MA, USA, 1986; pp. 19–34. [Google Scholar]
- Melbourne, B.A.; Hastings, A. Extinction risk depends strongly on factors contributing to stochasticity. Nature 2008, 454, 100–103. [Google Scholar] [CrossRef]
- King, J.L.; Jukes, T.H. Non-Darwinian Evolution: Most evolutionary change in proteins may be due to neutral mutations and genetic drift. Science 1969, 164, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Roemer, G.; Debenham, S.; Binns, M.; Garcelon, D.; Wayne, R.K. High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc. Natl. Acad. Sci. USA 2004, 101, 3490–3494. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Yeager, M. Natural selection at major histocompatibility complex loci of vertebrates. Annu. Rev. Genet. 1998, 32, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.X.; Zhang, B.Y.; Huang, K.; Ying, M.J.; Yan, J.B.; Niu, F.; Hu, H.Y.; Dunn, D.W.; Ren, Y.; Li, B.G.; et al. Balancing selection shapes population differentiation of major histocompatibility complex genes in wild golden snub-nosed monkeys. Curr. Zool. 2023, zoad043. [Google Scholar] [CrossRef]
- Herdegen, M.; Babik, W.; Radwan, J. Selective pressures on MHC class II genes in the guppy (Poecilia reticulata) as inferred by hierarchical analysis of population structure. J. Evol. Biol. 2014, 27, 2347–2359. [Google Scholar] [CrossRef]
- Rico, Y.; Morris-Pocock, J.; Zigouris, J.; Nocera, J.J.; Kyle, C.J. Lack of spatial immunogenetic structure among wolverine (Gulo gulo) populations suggestive of broad scale balancing selection. PLoS ONE 2015, 10, e0140170. [Google Scholar] [CrossRef]
- Yu, F.J.; Zhu, Y.; Xiong, T.Y.; Wan, Q.H.; Zhang, H.M. Balancing selection and recombination drive genetic variation at MHC class I genes in the giant panda. Sci. Bull. 2015, 60, 136–138. [Google Scholar] [CrossRef]
- Minias, P.; Whittingham, L.A.; Dunn, P.O. Coloniality and migration are related to selection on MHC genes in birds. Evolution 2017, 71, 432–441. [Google Scholar] [CrossRef]
- Zhang, P.; Huang, K.; Zhang, B.Y.; Dunn, D.W.; Chen, D.; Li, F.; Qi, X.G.; Li, B.G. High polymorphism in MHC-DRB genes in golden snub-nosed monkeys reveals balancing selection in small, isolated populations. BMC Evol. Biol. 2018, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Bleisch, W.V.; Richardson, M. Rhinopithecus bieti. The IUCN red list of threatened species 2020, e.T19597A17943738. Available online: https://www.iucnredlist.org/species/19597/17943738 (accessed on 10 June 2024).
- Long, Y.C.; Kirkpatrick, C.R.; Zhongtai; Xiaolin. Report on the distribution, population, and ecology of the Yunnan snub-nosed monkey (Rhinopithecus bieti). Primates 1994, 35, 241–250. [Google Scholar] [CrossRef]
- Zhu, S.X.; Li, L.; Slate, T.J.; Tang, H.X.; Wu, G.S.; Guo, H.Y.; Li, D.Y. The change in habitat quality for the Yunnan snub-nosed monkey from 1975 to 2022. Biology 2023, 12, 886. [Google Scholar] [CrossRef] [PubMed]
- Li, B.G.; Pan, R.L.; Oxnard, C.E. Extinction of snub-nosed monkeys in China during the past 400 years. Int. J. Primatol 2002, 23, 1227–1244. [Google Scholar] [CrossRef]
- Zhao, X.M.; Ren, B.P.; Garber, P.A.; Li, X.H.; Li, M. Impacts of human activity and climate change on the distribution of snub-nosed monkeys in China during the past 2000 years. Divers. Distrib. 2018, 24, 92–102. [Google Scholar] [CrossRef]
- Zhao, X.M.; Ren, B.P.; Li, D.Y.; Xiang, Z.F.; Garber, P.A.; Li, M. Effects of habitat fragmentation and human disturbance on the population dynamics of the Yunnan snub-nosed monkey from 1994 to 2016. PeerJ 2019, 7, e6633. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.P.; Li, D.Y.; Garber, P.A.; Li, M. Fission-fusion behavior in Yunnan snub-nosed monkeys (Rhinopithecus bieti) in Yunnan, China. Int. J. Primatol. 2012, 33, 1096–1109. [Google Scholar] [CrossRef]
- Xia, W.C.; Wang, F.; Wang, D.L.; Zeng, X.Q.; Yang, C.; Krzton, A.; Ren, B.P.; Li, D.Y. Dispersal patterns in Yunnan snub-nosed monkeys. Curr. Zool. 2022, 68, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.W.; Huo, S.; Zhong, T.; Xiang, Z.F.; Xiao, W.; Quan, R.C. Social organization of black-and-white snub-nosed monkeys (Rhinopithecus bieti) at Deqin, China. Am. J. Primatol. 2008, 70, 169–174. [Google Scholar] [CrossRef]
- Zhang, Y.; Ryder, O.A. Mitochondrial DNA sequence evolution and conservation relevance of snub-nosed langurs. Acta genet. Sin. 1997, 24, 116–121. [Google Scholar]
- Kuang, W.M.; Hu, J.Y.; Wu, H.; Fen, X.T.; Dai, Q.Y.; Fu, Q.M.; Xiao, W.; Frantz, L.; Roos, C.; Nadler, T.; et al. Genetic diversity, inbreeding level, and genetic load in endangered snub-nosed monkeys (Rhinopithecus). Front. Genet. 2020, 11, 615926. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Ren, B.P.; Wei, F.W.; Long, Y.C.; Hao, Y.L.; Li, M. Phylogeography and population structure of the yunnan snub-nosed monkey (Rhinopithecus bieti) inferred from mitochondrial control region dna sequence analysis. Mol. Ecol. 2007, 16, 3334–3349. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Ren, B.P.; Wu, R.D.; Zhao, L.; Hao, Y.L.; Wang, B.S.; Wei, F.W.; Long, Y.C.; Li, M. The effect of landscape features on population genetic structure in Yunnan snub-nosed monkeys (Rhinopithecus bieti) implies an anthropogenic genetic discontinuity. Mol. Ecol. 2009, 18, 3831–3846. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Wang, B.S.; Pan, Q.; Zhang, J.B.; Kumar, S.; Sun, X.Q.; Liu, Z.J.; Pan, H.J.; Lin, Y.; Liu, G.J.; et al. Whole-genome sequencing of the snub-nosed monkey provides insights into folivory and evolutionary history. Nat. Genet. 2014, 46, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, G.D.; Ruan, J.; Chen, Y.B.; Yang, C.P.; Cao, X.; Wu, H.; Liu, Y.H.; Du, Z.L.; Wang, X.P.; et al. Genomic analysis of snub-nosed monkeys (Rhinopithecus) identifies genes and processes related to high-altitude adaptation. Nat. Genet. 2016, 48, 947–952. [Google Scholar] [CrossRef]
- Liu, Z.J.; Ren, B.P.; Hao, Y.L.; Zhang, H.R.; Wei, F.W.; Li, M. Identification of 13 human microsatellite markers via cross-species amplification of fecal samples from Rhinopithecus biet. Int. J. Primatol. 2008, 29, 265–272. [Google Scholar] [CrossRef]
- Hao, Y.L.; Liu, Z.J.; Wu, H.; Ren, B.P.; Wei, F.W.; Li, M. Isolation and characterization of 11 microsatellite loci for the Sichuan snub-nosed monkey, Rhinopithecus roxellana. Conserv. Genet. 2007, 8, 1021–1024. [Google Scholar] [CrossRef]
- Taberlet, P.; Griffin, S.; Goossens, B.; Questiau, S.; Manceau, V.; Escaravage, N.; Waits, L.P.; Bouvet, J. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996, 24, 3189–3194. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.F.; Pan, H.J.; Liu, Z.J.; Li, M. Balancing selection and genetic drift at major histocompatibility complex class II genes in isolated populations of golden snub-nosed monkey (Rhinopithecus roxellana). BMC Evol. Biol. 2012, 12, 207. [Google Scholar] [CrossRef]
- Xu, H.L.; Wang, Y.T.; Cheng, A.C.; Yao, Y.F.; Ni, Q.Y.; Zen, W.; Bi, F.J.; Yang, Z.X.; Chen, X.Y. Polymorphism of MHC-DPB1 gene exon 2 in rhesus macaques (Macaca mulatta). Yichuan 2010, 32, 588–598. [Google Scholar]
- Zhang, P.; Zhang, B.Y.; Dunn, D.W.; Song, X.Y.; Huang, K.; Dong, S.X.; Niu, F.; Ying, M.J.; Zhang, Y.Y.; Shang, Y.X.; et al. Social and paternal female choice for male MHC genes in golden snub-nosed monkeys (Rhinopithecus roxellana). Mol. Ecol. 2023, 32, 3239–3256. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S.; Courtiol, A.; Mazzoni, C.J. MHC genotyping of non-model organisms using next-generation sequencing: A new methodology to deal with artefacts and allelic dropout. BMC Genom. 2013, 14, 542. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Zhang, P.; Dunn, D.W.; Wang, T.C.; Mi, R.; Li, B.G. Assigning alleles to different loci in amplifications of duplicated loci. Mol. Ecol. Resour. 2019, 19, 1240–1253. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2010, 19, 1099–1106. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP′007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Reche, P.A.; Reinherz, E.L. Sequence variability analysis of human class I and class II MHC molecules: Functional and structural correlates of amino acid polymorphisms. J. Mol. Biol. 2003, 331, 623–641. [Google Scholar] [CrossRef]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Nylander, J. MrModeltest, Version 2; Uppsala University: Uppsala, Sweden, 2004. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Altekar, G.; Dwarkadas, S.; Huelsenbeck, J.P.; Ronquist, F. Parallel metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 2004, 20, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Bontrop, R.E.; Dawkins, R.L.; Erlich, H.A.; Gyllensten, U.B.; Heise, E.R.; Jones, P.P.; Wakeland, E.K.; Watkins, D.I. Nomenclature for the major histocompatibility complexes of different species: A proposal. Immunogenetics 1990, 31, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Piertney, S.; Oliver, M. The evolutionary ecology of the major histocompatibility complex. Heredity 2006, 96, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Meng, X.H.; Liu, Z.J.; Chang, J.; Wang, B.S.; Li, M.Z.; Wengel, P.O.; Tian, S.L.; Wen, C.D.; Wang, Z.M.; et al. Population genomics reveals low genetic diversity and adaptation to hypoxia in snub-nosed monkeys. Mol. Biol. Evol. 2016, 33, 2670–2681. [Google Scholar] [CrossRef]
- Yang, M.Y.; Yang, Y.Q.; Cui, D.Y.; Fickenscher, G.; Zinner, D.; Roos, C.; Brameier, M. Population genetic structure of Guizhou snub-nosed monkeys (Rhinopithecus brelichi) as inferred from mitochondrial control region sequences, and comparison with R. roxellana and R. bieti. Am. J. Phys. Anthropol. 2012, 147, 1–10. [Google Scholar] [CrossRef]
- Luo, M.F.; Pan, H.J. MHC II DRB variation and trans-species polymorphism in the golden snub-nosed monkey (Rhinopithecus roxellana). Chin. Sci. Bull. 2013, 58, 2119–2127. [Google Scholar] [CrossRef]
- Song, X.Y.; Zhang, P.; Huang, K.; Chen, D.; Guo, S.T.; Qi, X.G.; He, G.; Pan, R.L.; Li, B.G. The influence of positive selection and trans-species evolution on DPB diversity in the golden snub-nosed monkeys (Rhinopithecus roxellana). Primates 2016, 57, 489–499. [Google Scholar] [CrossRef]
- Zhang, P.; Song, X.Y.; Dunn, D.W.; Huang, K.; Pan, R.L.; Chen, D.; Guo, S.T.; Qi, X.G.; He, G.; Li, B. Diversity at two genetic loci associated with the major histocompatibility complex in the golden snub-nosed monkey (Rhinopithecus roxellana). Biochem. Syst. Ecol. 2016, 68, 243–249. [Google Scholar] [CrossRef]
- Kuang, W.M.; Zinner, D.; Li, Y.; Yao, X.Q.; Roos, C.; Yu, L. Recent advances in genetics and genomics of snub-nosed monkeys (Rhinopithecus) and their implications for phylogeny, conservation, and adaptation. Genes 2023, 14, 985. [Google Scholar] [CrossRef] [PubMed]
- Díez-Del-Molino, D.; Sánchez-Barreiro, F.; Barnes, I.; Gilbert, M.T.P.; Dalén, L. Quantifying temporal genomic erosion in endangered species. Trends Eco. Evol. 2018, 33, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Solórzano-García, B.; White, J.M.; Shedden, A. Parasitism in heterogeneous landscapes: Association between conserved habitats and gastrointestinal parasites in populations of wild mammals. Acta Trop. 2023, 237, 1067751. [Google Scholar] [CrossRef] [PubMed]
- Tonteri, A.; Vasemägi, A.; Lumme, J.; Primmer, C.R. Beyond MHC: Signals of elevated selection pressure on Atlantic salmon (Salmo salar) immune-relevant loci. Mol. Ecol. 2010, 19, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Wang, S.Q.; Cheng, C.Y.; Zhang, J.Q.; Wang, S.P.; Hou, X.L.; Liu, X.; Yang, X.J.; Li, X.P. Latitudinal gradients in genetic diversity and natural selection at a highly adaptive gene in terrestrial mammals. Ecography 2021, 44, 206–218. [Google Scholar]
- Guernier, V.; Hochberg, M.E.; Guégan, J.F. Ecology drives the worldwide distribution of human diseases. PLoS Biol. 2004, 2, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Awadi, A.; Slimen, B.H.; Smith, S.; Knauer, F.; Makni, M.; Suchentrunk, F. Positive selection and climatic effects on MHC class II gene diversity in hares (Lepus capensis) from a steep ecological gradient. Sci. Rep. 2018, 8, 11514. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Sun, J.; Li, L.H.; Zhou, Y.; Fang, X.L.; Li, B.Y.; Guo, L.J.; Geng, Y.; Wang, C.P.; Huang, Z.P.; et al. Effects of provisioning on the activity budget and foraging strategies of black-and-white snub-nosed monkeys (Rhinopithecus bieti) in the Baima Snow Mountain Nature Reserve, Yunnan, China. Am. J. Primatol. 2023, 85, e23548. [Google Scholar] [CrossRef]
- Hou, R. Nutritional Ecology of the Golden Snub-Nosed Monkey (Rhinopithecus roxellana) and Its Adaption to Cold Environment. Ph.D. Thesis, Northwest University, Xi’an, China, 2018. [Google Scholar]
- Bollmer, J.L.; Hull, J.M.; Ernest, H.B.; Sarasola, J.H.; Parker, P.G. Reduced MHC and neutral variation in the Galápagos hawk, an island endemic. BMC Evol. Biol. 2011, 11, 143. [Google Scholar] [CrossRef]
- Blais, J.; Rico, C.; van Oosterhout, C.; Cable, J.; Turner, G.F.; Bernatchez, L. MHC adaptive divergence between closely related and sympatric african cichlids. PLoS ONE 2007, 2, e734. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.C.; Allendorf, F.; Daugherty, C.H. Genetic diversity and differentiation at MHC genes in island populations of tuatara (Sphenodon spp.). Mol. Ecol. 2010, 19, 3894–3908. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, C.; Richard, M.; Garnier, S.; Chastel, O.; Julliard, R.; Zoorob, R.; Sorci, G. Diversifying selection on MHC class I in the house sparrow (Passer domesticus). Mol. Ecol. 2009, 18, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shen, H.; Wang, H.; Zhao, M.; Luo, Z.; Wu, H. Diversifying selection is the dominant factor affecting the geographical variation of MHC class II genes in the Omei tree frog. J. Zool. 2016, 300, 197–204. [Google Scholar] [CrossRef]
- Ekblom, R.; Saether, S.A.; Jacobsson, P.; Fiske, P.; Sahlman, T.; Grahn, M.; Kålås, J.A.; Höglund, J. Spatial pattern of MHC class II variation in the great snipe (Gallinago media). Mol. Ecol. 2007, 16, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Spurgin, L.G.; Richardson, D.S. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc. Biol. Sci. 2010, 277, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Dearborn, D.C.; Warren, S.; Hailer, F. Meta-analysis of major histocompatibility complex (MHC) class IIA reveals polymorphism and positive selection in many vertebrate species. Mol. Ecol. 2022, 31, 6390–6406. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W. Balancing selection and MHC. Genetica 1998, 104, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, L.; Landry, C. MHC studies in nonmodel vertebrates: What have we learned about natural selection in 15 years? J. Evol. Biol. 2003, 16, 363–377. [Google Scholar] [CrossRef]
- Slade, J.W.G.; Watson, M.J.; MacDougall-Shackleton, E.A. “Balancing” balancing selection? Assortative mating at the major histocompatibility complex despite molecular signatures of balancing selection. Ecol. Evol. 2019, 9, 5146–5157. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Liu, Y.; Song, M.J.; Lai, J.S.; Sun, J.H.; Gong, Q. Molecular polymorphism and expression of MHC I α, II α, II β and II invariant chain in the critically endangered Dabry’s sturgeon (Acipenser dabryanus). Dev. Comp. Immunol. 2020, 103, 103494. [Google Scholar] [CrossRef] [PubMed]
- Lau, Q.; Igawa, T.; Komaki, S.; Satta, Y. Characterisation of major histocompatibility complex class I genes in Japanese Ranidae frogs. Immunogenetics 2016, 68, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Stiebens, V.A.; Merino, S.E.; Chain, F.J.J.; Eizaguirre, C. Evolution of MHC class I genes in the endangered loggerhead sea turtle (Caretta caretta) revealed by 454 amplicon sequencing. BMC Evol. Biol. 2013, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Winternitz, J.; Chakarov, N.; Rinaud, T.; Ottensmann, M.; Krüger, O. High functional allelic diversity and copy number in both MHC classes in the common buzzard. BMC Ecol. Evol. 2023, 23, 24. [Google Scholar] [CrossRef] [PubMed]
- Stefanović, M.; Ćirović, D.; Bogdanović, N.; Knauer, F.; Heltai, M.; Szabó, L.; Lanszki, J.; Zhelev, C.D.; Schaschl, H.; Suchentrunk, F. Positive selection on the MHC class II DLA-DQA1 gene in golden jackals (Canis aureus) from their recent expansion range in Europe and its effect on their body mass index. BMC Ecol. Evol. 2021, 21, 122. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, E.; Kappeler, P.M.; Brameier, M.; Demeler, J.; Kraus, C.; Rakotoniaina, J.H.; Hämäläinen, A.M.; Huchard, E. Shared evolutionary origin of major histocompatibility complex polymorphism in sympatric lemurs. Mol. Ecol. 2017, 26, 5629–5645. [Google Scholar] [CrossRef]
- Talarico, L.; Babik, W.; Marta, S.; Pietrocini, V.; Mattoccia, M. MHC structuring and divergent allele advantage in a urodele amphibian: A hierarchical multi-scale approach. Heredity 2019, 123, 593–607. [Google Scholar] [CrossRef]
- Schierup, M.H.; Vekemans, X.; Charlesworth, D. The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet. Res. 2000, 76, 51–62. [Google Scholar] [CrossRef] [PubMed]
- van Oosterhout, C.; Joyce, D.A.; Cummings, S.M.; Blais, J.; Barson, N.J.; Ramnarine, I.W.; Mohammed, R.S.; Persad, N.; Cable, J. Balancing selection, random genetic drift, and genetic variation at the major histocompatibility complex in two wild populations of guppies (Poecilia reticulata). Evolution 2006, 60, 2562–2574. [Google Scholar] [CrossRef]
- Huang, W.; Dicks, K.L.; Hadfield, J.D.; Johnston, S.E.; Ballingall, K.T.; Pemberton, J.M. Contemporary selection on MHC genes in a free-living ruminant population. Ecol. Lett. 2022, 25, 828–838. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Locus | AR | AE | HO | HE | PIC | FIS | Null | HWE |
|---|---|---|---|---|---|---|---|---|
| GM108 | 5 | 3.386 | 0.771 | 0.712 | 0.652 | −0.084 | −0.045 | NS |
| D17S1290 | 4 | 1.554 | 0.354 | 0.360 | 0.335 | 0.017 | −0.002 | NS |
| GM109 | 9 | 2.794 | 0.667 | 0.649 | 0.608 | −0.028 | −0.010 | NS |
| D11S2002 | 7 | 4.455 | 0.652 | 0.784 | 0.741 | 0.170 | 0.079 | *** |
| D1S533 | 6 | 3.949 | 0.792 | 0.755 | 0.708 | −0.05 | −0.038 | NS |
| D6S474 | 3 | 1.349 | 0.250 | 0.262 | 0.242 | 0.045 | 0.003 | NS |
| D1s207 | 5 | 2.100 | 0.417 | 0.529 | 0.465 | 0.215 | 0.134 | NS |
| GM214 | 7 | 2.636 | 0.604 | 0.627 | 0.567 | 0.037 | 0.026 | NS |
| D6S493 | 3 | 1.647 | 0.438 | 0.407 | 0.363 | −0.075 | −0.045 | NS |
| Average | 5.444 | 2.705 | 0.549 | 0.565 | 0.520 | −0.047 | 0.011 |
| Locus | A | Pi | AE | HO | HE | PIC | FIS | HWE |
|---|---|---|---|---|---|---|---|---|
| DQA1 | 2 | 0.146 | 1.900 | 0.521 | 0.474 | 0.362 | −0.070 | NS |
| DQB1 | 2 | 0.107 | 1.999 | 0.604 | 0.500 | 0.375 | −0.227 | NS |
| DRB1 | 2 | 0.074 | 1.999 | 0.438 | 0.500 | 0.375 | −0.054 | NS |
| DRB5 | 2 | 0.079 | 1.999 | 0.438 | 0.500 | 0.375 | 0.117 | NS |
| DPB1 | 3 | 0.106 | 2.121 | 0.563 | 0.528 | 0.426 | −0.023 | NS |
| Average | 2.200 | 0.102 | 2.003 | 0.513 | 0.500 | 0.383 | −0.051 |
| Locus | Model | #p | Log Likelihood | Estimate Parameters | Positively Selected Sites |
|---|---|---|---|---|---|
| DQA1 | M0 (one ratio) | 1 | −458.470 | ω0 = 0.749 | None |
| M1a (nearly neutral) | 2 | −458.194 | p0 = 0.250 (p1 = 0.750) | Not allowed | |
| M2a (positive selection) | 4 | −457.457 | p0 = 0.965, p1 = 0.000 (p2 = 0.035) ω2 = 17.868 | 54F, 62G | |
| M3 (discrete) | 5 | −457.457 | p0 = 0.000, p1 = 0.965 (p2 = 0.035) ω1 = 0.678, ω2 = 17.868 | 54F, 62G | |
| M7 (beta) | 2 | −458.209 | p = 0.039, q = 0.014 | Not allowed | |
| M8 (beta and omega) | 4 | −457.457 | p0 = 0.965 (p1 = 0.035) p = 99.000, q = 46.920, ωs = 17.878 | 54F, 62G | |
| DPB1 | M0 (one ratio) | 1 | −525.044 | ω0 = 0.328 | None |
| M1a (nearly neutral) | 2 | −523.208 | p0 = 0.548 (p1 = 0.452) | Not allowed | |
| M2a (positive selection) | 4 | −523.208 | p0 = 0.548, p1 = 0.377 (p2 = 0.075) ω2 = 1.000 | 60L | |
| M3 (discrete) | 5 | −523.197 | p0 = 0.535, p1 = 0.226 (p2 = 0.239) ω1 = 0.922, ω2 = 0.922 | Not allowed | |
| M7 (beta) | 2 | −523.218 | p = 0.024, q = 0.030 | Not allowed | |
| M8 (beta and omega) | 4 | −523.218 | p0 = 0.999 (p1 = 0.000) p = 0.024, q = 0.031, ωs = 2.799 | 60L | |
| DQB1 | M0 (one ratio) | 1 | −459.831 | ω0 = 0.731 | None |
| M1a (nearly neutral) | 2 | −457.833 | p0 = 0.494 (p1 = 0.506) | Not allowed | |
| M2a (positive selection) | 4 | −454.253 | p0 = 0.947, p1 = 0.000 (p2 = 0.053) ω2 = 52.061 | 21L, 52S, 55Y, 75R | |
| M3 (discrete) | 5 | −454.253 | p0 = 0.000, p1 = 0.947 (p2 = 0.053) ω1 = 0.530, ω2 = 52.061 | 21L, 52S, 55Y, 75R | |
| M7 (beta) | 2 | −457.834 | p = 0.005, q = 0.005 | Not allowed | |
| M8 (beta and omega) | 4 | −454.254 | p0 = 0.947 (p1 = 0.053) p = 99.000, q = 87.548, ωs = 52.068 | 21L, 52S, 55Y, 75R | |
| DRB1 | M0 (one ratio) | 1 | −427.018 | ω0 = 0.734 | None |
| M1a (nearly neutral) | 2 | −424.528 | p0 = 0.615 (p1 = 0.385) | Not allowed | |
| M2a (positive selection) | 4 | −419.12 | p0 = 0.000, p1 = 0.931 (p2 = 0.069) ω2 = 161.196 | 5Q, 6A, 7K, 26I, 27H, 32N, 42F, 44A, 59Q, 66E, 73Y, 81F, 82D | |
| M3 (discrete) | 5 | −418.946 | p0 = 0.050 p1 = 0.879 (p2 = 0.071) ω1 = 0.581, ω2 = 96.582 | 5Q, 6A, 7K, 73Y, 81F | |
| M7 (beta) | 2 | −424.533 | p = 0.005, q = 0.008 | Not allowed | |
| M8 (beta and omega) | 4 | −418.946 | p0 = 0.929 (p1 = 0.071) p = 99.000, q = 71.380, ωs = 96.604 | 5Q, 6A, 7K, 26I, 27H, 32N, 42F, 44A, 59Q, 66E, 73Y, 81F, 82D | |
| DRB5 | M0 (one ratio) | 1 | −411.081 | ω0 = 0.349 | None |
| M1a (nearly neutral) | 2 | −408.087 | p0 = 0.703 (p1 = 0.297) | Not allowed | |
| M2a (positive selection) | 4 | −407.351 | p0 = 0.792, p1 = 0.000 (p2 = 0.208) ω2 = 2.596 | 23E, 32F, 69R | |
| M3 (discrete) | 5 | −407.351 | p0 = 0.624, p1 = 0.168 (p2 = 0.208) ω1 = 0.000, ω2 = 2.596 | 4Q, 8L, 20Q, 23E, 25Y, 32F, 42F, 46S, 52E, 55N, 69R | |
| M7 (beta) | 2 | −408.088 | p = 0.005, q = 0.012 | Not allowed | |
| M8 (beta and omega) | 4 | −407.351 | p0 = 0.792 (p1 = 0.208) p = 0.005, q = 80.070, ωs = 2.596 | 23E, 32F, 69R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Song, C.; Liang, J.; La, Y.; Lai, J.; Pan, R.; Huang, Z.; Li, B.; Zhang, P. Moderate Genetic Diversity of MHC Genes in an Isolated Small Population of Black-and-White Snub-Nosed Monkeys (Rhinopithecus bieti). Animals 2024, 14, 2276. https://doi.org/10.3390/ani14152276
Yan J, Song C, Liang J, La Y, Lai J, Pan R, Huang Z, Li B, Zhang P. Moderate Genetic Diversity of MHC Genes in an Isolated Small Population of Black-and-White Snub-Nosed Monkeys (Rhinopithecus bieti). Animals. 2024; 14(15):2276. https://doi.org/10.3390/ani14152276
Chicago/Turabian StyleYan, Jibing, Chunmei Song, Jiaqi Liang, Yanni La, Jiandong Lai, Ruliang Pan, Zhipang Huang, Baoguo Li, and Pei Zhang. 2024. "Moderate Genetic Diversity of MHC Genes in an Isolated Small Population of Black-and-White Snub-Nosed Monkeys (Rhinopithecus bieti)" Animals 14, no. 15: 2276. https://doi.org/10.3390/ani14152276