
Natural Foraging Selection and Gut Microecology of Two Subterranean Rodents from the Eurasian Steppe in China

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
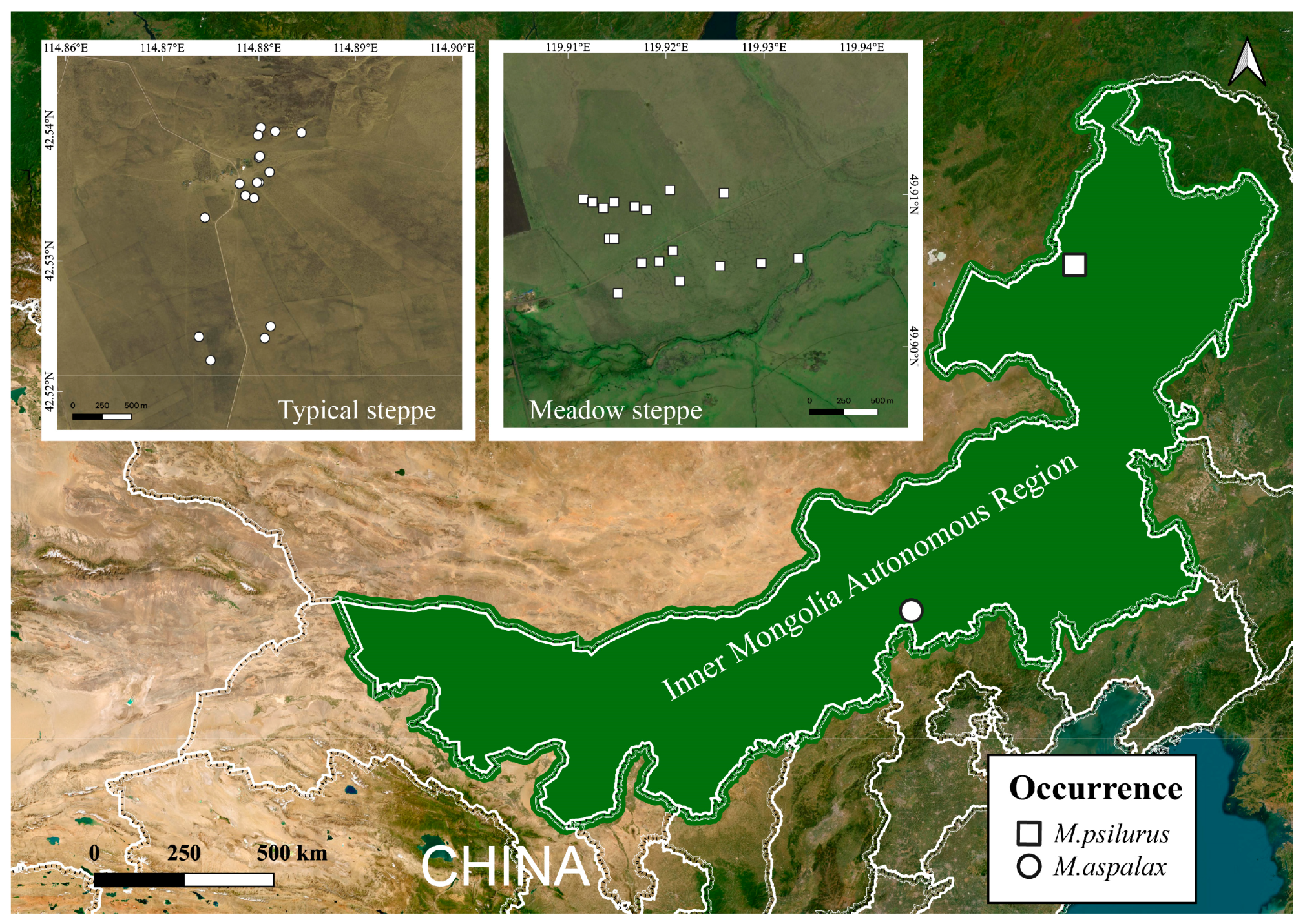
2.1. Sample Collection
2.2. DNA Extraction and Sequencing of Cecum and Stomach Contents
2.3. Bioinformatics Analysis
3. Results
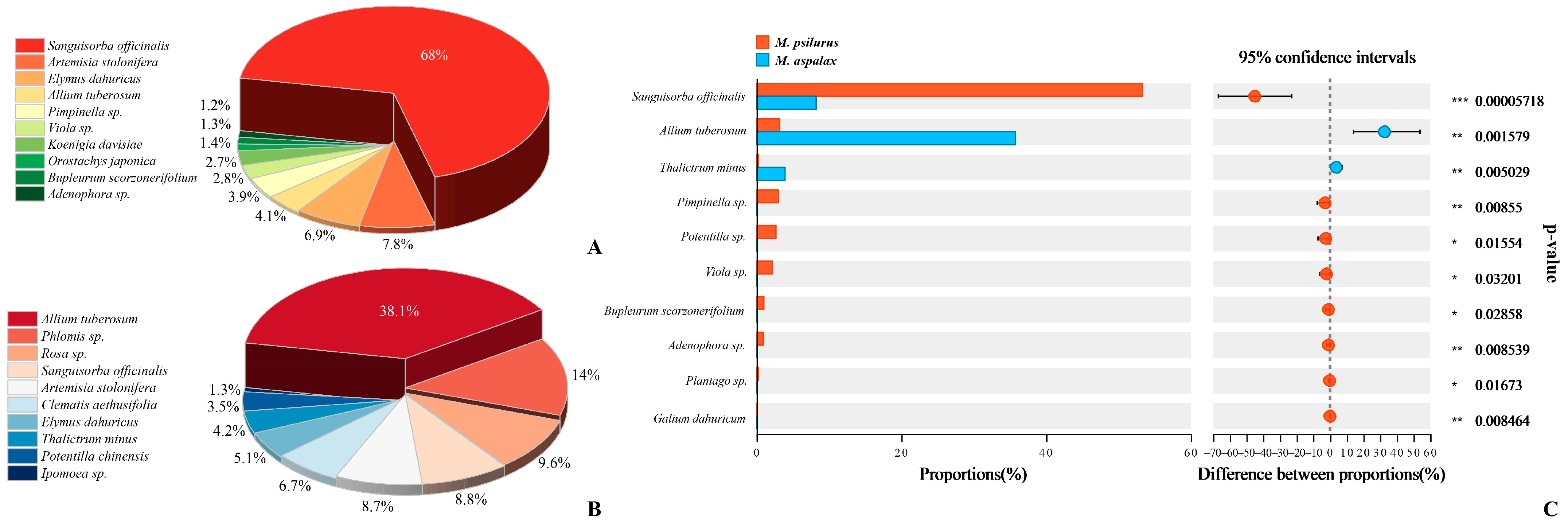
3.1. Analysis of Food Composition and Dietary Differences
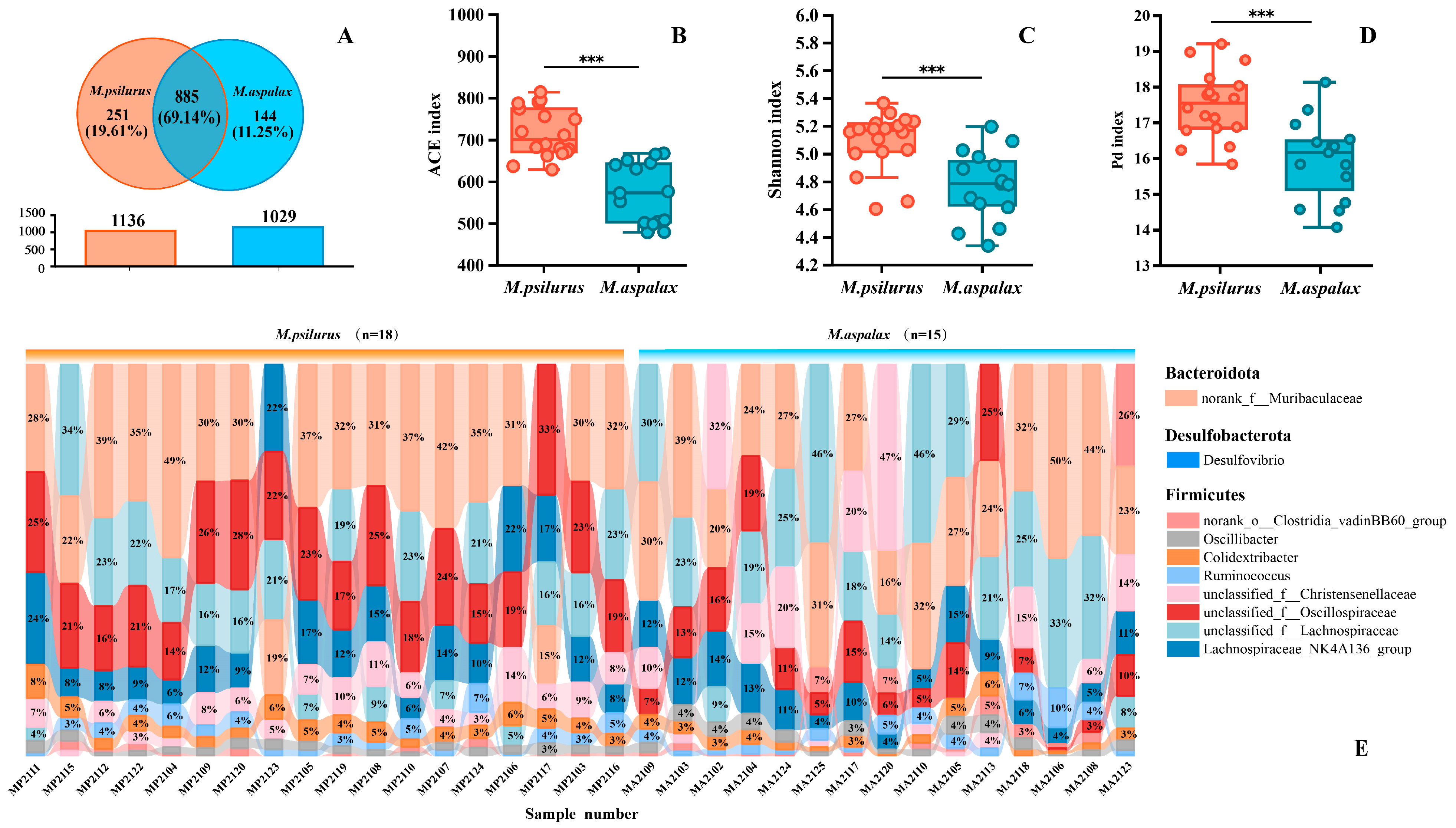
3.2. Sequencing Information and Gut Microbial α-Diversity
3.3. Analysis of the Association between Food Types and Gut Microorganisms
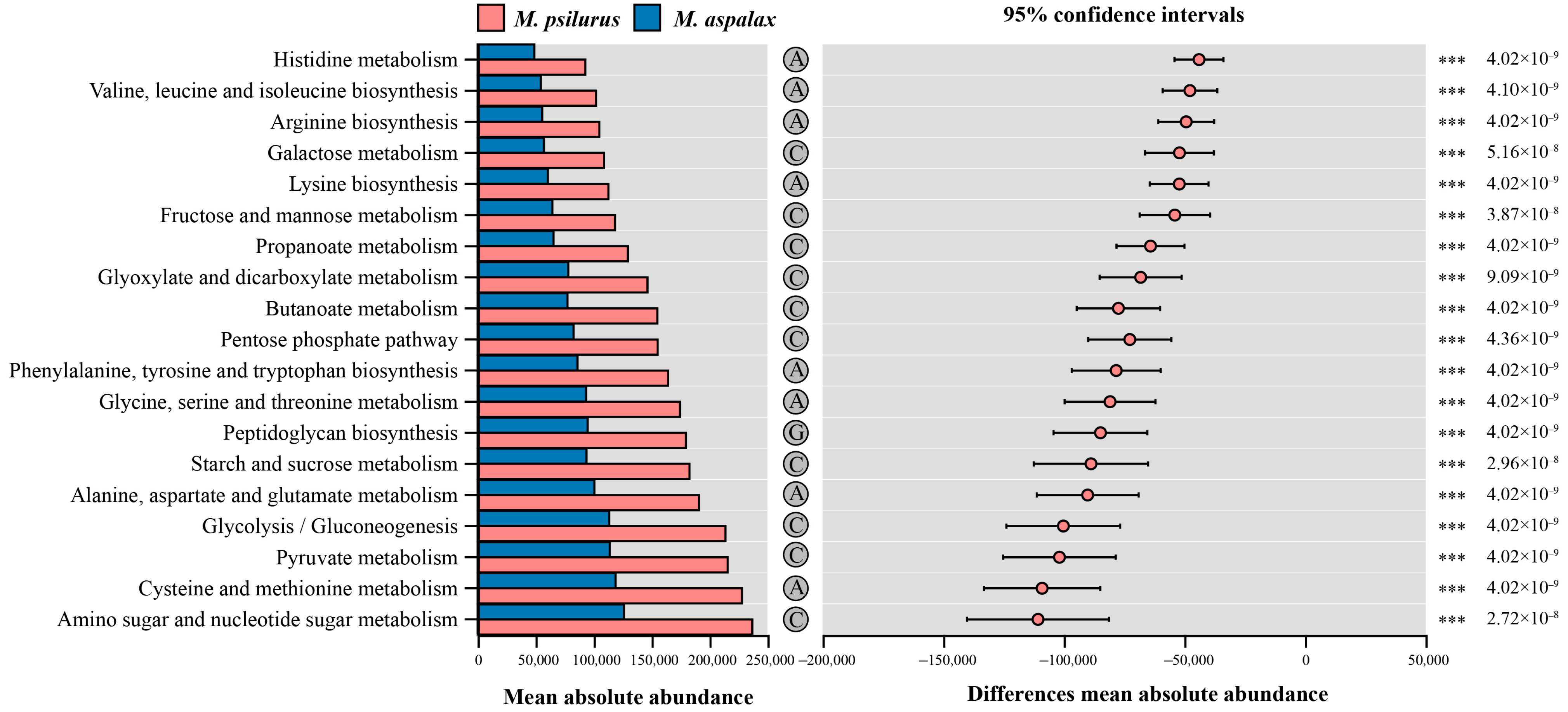
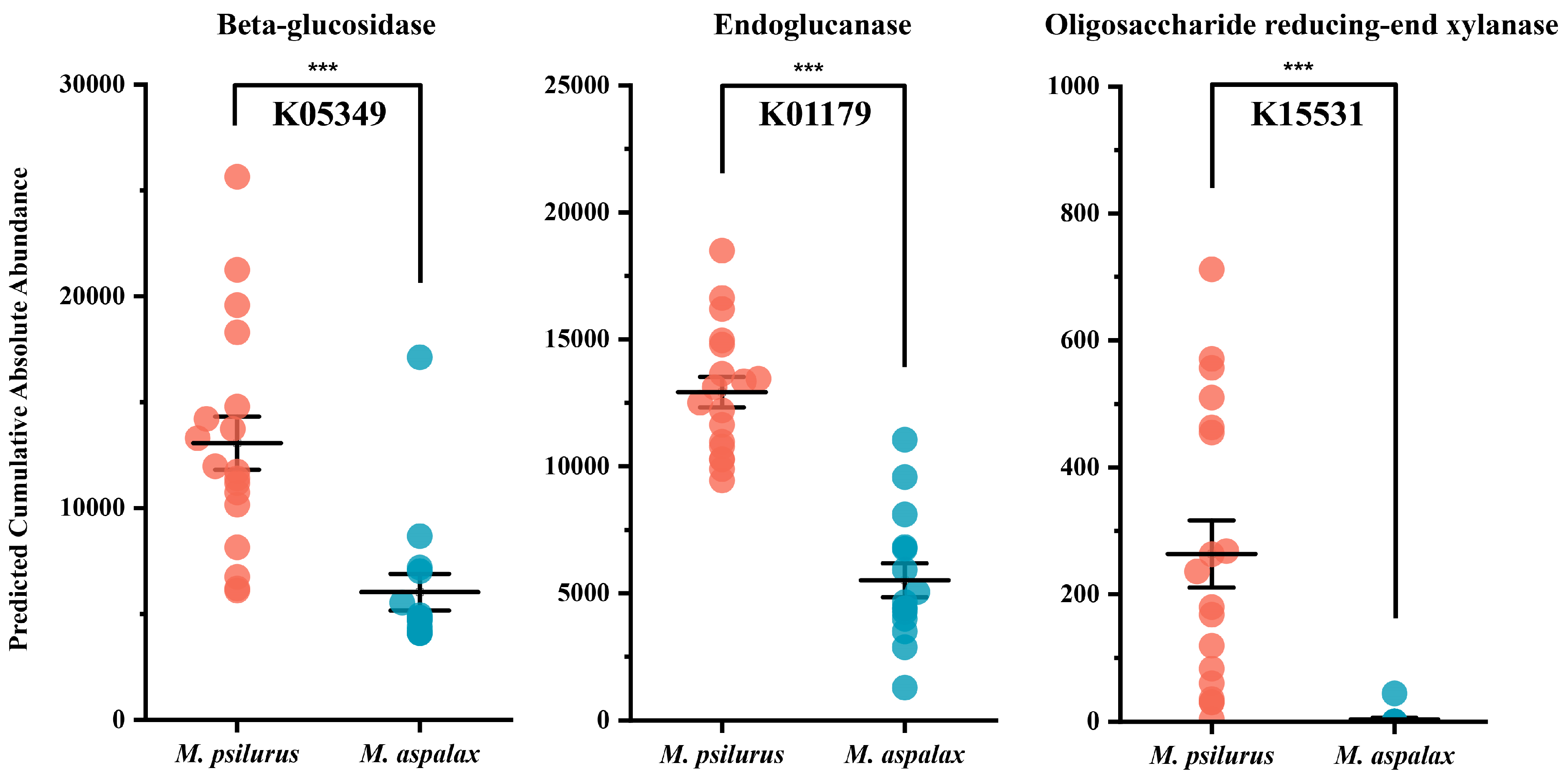
3.4. Indicator Genera and Functional Prediction of Two Zokor Species’ Gut Bacteria
3.5. Gut Bacteria Associated with Cellulose Degradation
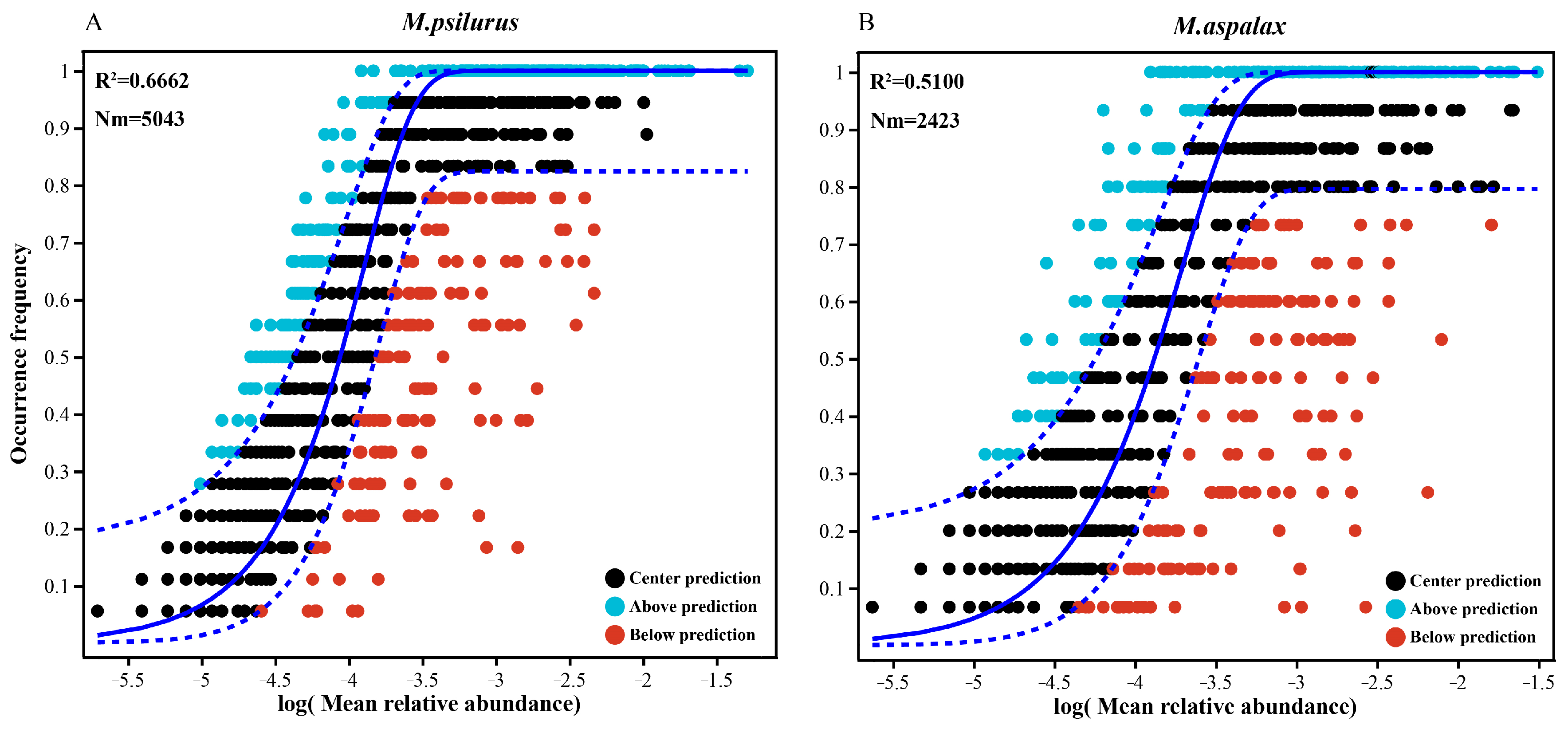
3.6. Neutral Community Modelling (NCM) of Gut Flora
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Best, N.; Rolle-Kampczyk, U.; Schaap, F.G.; Basic, M.; Olde Damink, S.W.M.; Bleich, A.; Savelkoul, P.H.M.; von Bergen, M.; Penders, J.; Hornef, M.W. Bile Acids Drive the Newborn’s Gut Microbiota Maturation. Nat. Commun. 2020, 11, 3692. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and Evolutionary Forces Shaping Microbial Diversity in the Human Intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The Role of the Gut Microbiota in Nutrition and Health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Baxter, N.T.; Lesniak, N.A.; Sinani, H.; Schloss, P.D.; Koropatkin, N.M. The Glucoamylase Inhibitor Acarbose Has a Diet-Dependent and Reversible Effect on the Murine Gut Microbiome. mSphere 2019, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Correa, M.A.; Matusovsky, B.; Brackney, D.E.; Steven, B. Generation of Axenic Aedes aegypti Demonstrate Live Bacteria Are Not Required for Mosquito Development. Nat. Commun. 2018, 9, 4464. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, T.; Zeng, Y.; Pei, P.; Liu, Y.; Jia, W.; Zhao, H.; Bi, M.; Wang, S. Sodium Butyrate Mediates Histone Crotonylation and Alleviated Neonatal Rats Hypoxic–Ischemic Brain Injury through Gut–Brain Axis. Front. Microbiol. 2022, 13, 993146. [Google Scholar] [CrossRef] [PubMed]
- Angoa-Pérez, M.; Zagorac, B.; Francescutti, D.M.; Winters, A.D.; Greenberg, J.M.; Ahmad, M.M.; Manning, S.D.; Gulbransen, B.D.; Theis, K.R.; Kuhn, D.M. Effects of a High Fat Diet on Gut Microbiome Dysbiosis in a Mouse Model of Gulf War Illness. Sci. Rep. 2020, 10, 9529. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.-H.; Chou, C.-H.; Huang, T.-Y.; Wang, H.-L.; Chien, P.-J.; Chang, W.-W.; Lee, H.-T. Heat-Killed Lactobacilli Preparations Promote Healing in the Experimental Cutaneous Wounds. Cells 2021, 10, 3264. [Google Scholar] [CrossRef]
- Kang, P.; Pan, Y.; Pan, Y.; Hu, J.; Zhao, T.; Zhang, Y.; Ding, X.; Yan, X. A Comparison of Microbial Composition under Three Tree Ecosystems Using the Stochastic Process and Network Complexity Approaches. Front. Microbiol. 2022, 13, 1018077. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, L.; Ding, J.; Dai, R.; He, C.; Xu, K.; Luo, L.; Xiao, L.; Zheng, Y.; Han, C.; et al. Intestinal Microbiota and Host Cooperate for Adaptation as a Hologenome. mSystems 2022, 7, e01261-21. [Google Scholar] [CrossRef]
- Kim, J.-S.; Kang, S.W.; Lee, J.H.; Park, S.-H.; Lee, J.-S. The Evolution and Competitive Strategies of Akkermansia muciniphila in Gut. Gut Microbes 2022, 14, 2025017. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Mazel, F.; Alm, E.J. Co-Evolution and Co-Speciation of Host-Gut Bacteria Systems. Cell Host Microbe 2020, 28, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, H.; Jiang, F.; Liu, D.; Hou, Y.; Chi, X.; Qin, W.; Song, P.; Cai, Z.; Zhang, T. Comparative Analysis of Gut Microbial Composition and Functions in Przewalski’s Gazelle (Procapra przewalskii) from Various Habitats. Front. Microbiol. 2022, 13, 913358. [Google Scholar] [CrossRef] [PubMed]
- Doyle, C.J.; Gleeson, D.; O’Toole, P.W.; Cotter, P.D. Impacts of Seasonal Housing and Teat Preparation on Raw Milk Microbiota: A High-Throughput Sequencing Study. Appl. Environ. Microbiol. 2016, 83, e02694-16. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.; Bird, A. The Impact of Diet and Lifestyle on Gut Microbiota and Human Health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, H.; Feng, Z.; Cao, J.; Fang, C.; Xu, X.; Zhao, L.; Shen, J. Development of Human Breast Milk Microbiota-Associated Mice as a Method to Identify Breast Milk Bacteria Capable of Colonizing Gut. Front. Microbiol. 2017, 8, 1242. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-Y.; Han, B.; Deng, X.; Deng, S.-Y.; Zhang, Y.-Y.; Shen, P.-X.; Hui, T.; Chen, R.-H.; Li, X.; Zhang, Y. Pomegranate Peel Extract Ameliorates the Severity of Experimental Autoimmune Encephalomyelitis via Modulation of Gut Microbiota. Gut Microbes 2020, 12, 1857515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhou, Y.; Wu, Y.; Zhou, K.; Liu, A.; Yang, F.; Zhang, W. Prevalence and Genetic Characterization of Two Mitochondrial Gene Sequences of Strobilocercus Fasciolaris in the Livers of Brown Rats (Rattus norvegicus) in Heilongjiang Province in Northeastern China. Front. Cell. Infect. Microbiol. 2020, 10, 588107. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Álvarez, S.; Karlsson, E.; Ryder, O.A.; Lindblad-Toh, K.; Crawford, A.J. How to Make a Rodent Giant: Genomic Basis and Tradeoffs of Gigantism in the Capybara, the World’s Largest Rodent. Mol. Biol. Evol. 2020, 38, 1715–1730. [Google Scholar] [CrossRef]
- Gettings, S.M.; Maxeiner, S.; Tzika, M.; Cobain, M.R.D.; Ruf, I.; Benseler, F.; Brose, N.; Krasteva-Christ, G.; Vande Velde, G.; Schönberger, M.; et al. Two Functional Epithelial Sodium Channel Isoforms Are Present in Rodents despite Pronounced Evolutionary Pseudogenization and Exon Fusion. Mol. Biol. Evol. 2021, 38, 5704–5725. [Google Scholar] [CrossRef]
- Cerling, T.E.; Andanje, S.A.; Blumenthal, S.A.; Brown, F.H.; Chritz, K.L.; Harris, J.M.; Hart, J.A.; Kirera, F.M.; Kaleme, P.; Leakey, L.N.; et al. Dietary Changes of Large Herbivores in the Turkana Basin, Kenya from 4 to 1 Ma. Proc. Natl. Acad. Sci. USA 2015, 112, 11467–11472. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Araujo, M.; Taş, N.; Alcantara-Hernandez, R.J.; Gaona, O.; Schondube, J.E.; Medellin, R.A.; Jansson, J.K.; Falcon, L.I. Phyllostomid Bat Microbiome Composition Is Associated to Host Phylogeny and Feeding Strategies. Front. Microbiol. 2015, 6, 447. [Google Scholar] [CrossRef] [PubMed]
- Higginson, A.D.; McNamara, J.M. An Adaptive Response to Uncertainty Can Lead to Weight Gain during Dieting Attempts. Evol. Med. Public Health 2016, 2016, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, T.; Beasley, D.E.; Heděnec, P.; Xiao, Z.; Zhang, S.; Li, J.; Lin, Q.; Li, X. Diet Diversity Is Associated with Beta but Not Alpha Diversity of Pika Gut Microbiota. Front. Microbiol. 2016, 7, 1169. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, G.; Bêty, J.; Cadieux, M.-C.; Legagneux, P.; Doiron, M.; Chevallier, C.; Lai, S.; Tarroux, A.; Berteaux, D. Long-Term Monitoring at Multiple Trophic Levels Suggests Heterogeneity in Responses to Climate Change in the Canadian Arctic Tundra. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120482. [Google Scholar] [CrossRef] [PubMed]
- Schaffers, A.P.; Raemakers, I.P.; Sýkora, K.V.; ter Braak, C.J.F. Arthropod Assemblages Are Best Predicted by Plant Species Composition. Ecology 2008, 89, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Mieke, V.D.H.; Bunce, M.; Dixon, K.W.; Fernandes, K.; Majer, J.; Wardell-Johnson, G.; White, N.E.; Nevill, P. Evaluating Restoration Trajectories Using DNA Metabarcoding of Ground-Dwelling and Airborne Invertebrates and Associated Plant Communities. Mol. Ecol. 2022, 31, 2172–2188. [Google Scholar] [CrossRef]
- Côté, S.D.; Rooney, T.P.; Tremblay, J.-P.; Dussault, C.; Waller, D.M. Ecological Impacts of Deer Overabundance. Annu. Rev. Ecol. Evol. Syst. 2004, 35, 113–147. [Google Scholar] [CrossRef]
- Estes, J.A.; Terborgh, J.; Brashares, J.S.; Power, M.E.; Berger, J.; Bond, W.J.; Carpenter, S.R.; Essington, T.E.; Holt, R.D.; Jackson, J.B.C.; et al. Trophic Downgrading of Planet Earth. Science 2011, 333, 301–306. [Google Scholar] [CrossRef]
- Zhou, Q.H.; Huang, Z.H.; Wei, H.; Huang, C.M.; Huang, Z.H.; Wei, H.; Huang, C.M. Variations in Diet Composition of Sympatric Trachypithecus francoisi and Macaca assamensis in the Limestone Habitats of Nonggang, China. Zool. Res. 2018, 39, 284–290. [Google Scholar] [CrossRef]
- Cao, H.Q.; Yang, X.W.; Peng, C.C.; Wang, Y.Y.; Guo, Q.Y.; Su, H.J. Winter Dietary Analysis Reveals the Foraging Differences of Wild Boar (Sus scrofa) in Different Regions of a Karst Mountainous Area. Animals 2023, 13, 727. [Google Scholar] [CrossRef]
- Mallott, E.K.; Garber, P.A.; Malhi, R.S. TrnL Outperforms RbcL as a DNA Metabarcoding Marker When Compared with the Observed Plant Component of the Diet of Wild White-Faced Capuchins (Cebus capucinus, Primates). PLoS ONE 2018, 13, e0199556. [Google Scholar] [CrossRef] [PubMed]
- Shrader, A.M.; Owen-Smith, N.; Ogutu, J.O. How a Mega-Grazer Copes with the Dry Season: Food and Nutrient Intake Rates by White Rhinoceros in the Wild. Funct. Ecol. 2006, 20, 376–384. [Google Scholar] [CrossRef]
- Hou, R.; Chapman, C.A.; Rothman, J.M.; Zhang, H.; Huang, K.; Guo, S.; Li, B.; Raubenheimer, D. The Geometry of Resource Constraint: An Empirical Study of the Golden Snub-Nosed Monkey. J. Anim. Ecol. 2021, 90, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Persson, A.; Stenberg, M. Linking Patch-Use Behavior, Resource Density, and Growth Expectations in Fish. Ecology 2006, 87, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Persson, L. Food Consumption and the Significance of Detritus and Algae to Intraspecific Competition in Roach Rutilus Rutilus in a Shallow Eutrophic Lake. Oikos 1983, 41, 118. [Google Scholar] [CrossRef]
- Cox, P.G.; Rayfield, E.J.; Fagan, M.J.; Herrel, A.; Pataky, T.C.; Jeffery, N. Functional Evolution of the Feeding System in Rodents. PLoS ONE 2012, 7, e36299. [Google Scholar] [CrossRef] [PubMed]
- Verde Arregoitia, L.D.; D’Elía, G. Classifying Rodent Diets for Comparative Research. Mammal Rev. 2020, 51, 51–65. [Google Scholar] [CrossRef]
- Butkauskas, D.; Starodubaitė, M.; Potapov, M.; Potapova, O.; Abramov, S.; Litvinov, Y. Phylogenetic Relationships between Zokors Myospalax (Mammalia, Rodentia) Determined on the Basis of Morphometric and Molecular Analyses. Proc. Latv. Acad. Sci. Sect. B Nat. Exact Appl. Sci. 2020, 74, 25–34. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, S.; Cai, Z.; Kuang, Z.; Wan, N.; Wang, Y.; Mao, L.; An, X.; Li, F.; Feng, T.; et al. Genomic Insights into Zokors’ Phylogeny and Speciation in China. Proc. Natl. Acad. Sci. USA 2022, 119, e2121819119. [Google Scholar] [CrossRef]
- Wei, F.; Yang, Q.; Wu, Y.; Jiang, X.; Liu, S.; Li, B.; Yang, G.; Li, M.; Zhou, J.; Li, S.; et al. Catalogue of Mammals in China. Acta Theriol. Sin. 2021, 41, 487–501. [Google Scholar] [CrossRef]
- Wang, N. Chromosomal Rearrangements and Speciation of Subterranean Myospalax in China. Master’s Thesis, Lanzhou University, Lanzhou, China, 2023; pp. 45–46. [Google Scholar]
- Puzachenko, A.; Pavlenko, M.; Korablev, V.; Tsvirka, M. Karyotype, Genetic and Morphological Variability in North China Zokor, Myospalax Psilurus (Rodentia, Spalacidae, Myospalacinae). Russ. J. Theriol. 2014, 13, 27–46. [Google Scholar] [CrossRef]
- Manduhu; Yuan, S.; Yang, S.; Ji, Y.; Chaoketu; Wei, J.; Fu, H.; Wu, X. Activity Pattern of Transbaikal Zokor (Myospalax psilurus) and Its Relationship with Soil Temperature and Humidity. Acta Theriol. Sin. 2021, 41, 441. [Google Scholar] [CrossRef]
- Zhang, T.; Lei, M.; Zhou, H.; Chen, Z.; Shi, P. Phylogenetic Relationships of the Zokor Genus Eospalax (Mammalia, Rodentia, Spalacidae) Inferred from Whole-Genome Analyses, with Description of a New Species Endemic to Hengduan Mountains. Zool. Res. 2022, 43, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sha, Y.; Lv, W.; Cao, G.; Guo, X.; Pu, X.; Wang, J.; Li, S.; Hu, J.; Luo, Y. Multi-Omics Reveals That the Rumen Transcriptome, Microbiome, and Its Metabolome Co-Regulate Cold Season Adaptability of Tibetan Sheep. Front. Microbiol. 2022, 13, 859601. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Zeng, D.; Qu, L.J.; Wang, Z.; Ning, Z.H. Multi-Enzyme Supplementation Modifies the Gut Microbiome and Metabolome in Breeding Hens. Front. Microbiol. 2021, 12, 711905. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.W.; Thieme, N.; Huberman, L.B.; Dietschmann, A.; Kowbel, D.J.; Lee, J.; Calhoun, S.; Singan, V.R.; Lipzen, A.; Xiong, Y.; et al. The Regulatory and Transcriptional Landscape Associated with Carbon Utilization in a Filamentous Fungus. Proc. Natl. Acad. Sci. USA 2020, 117, 6003–6013. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, C.; Li, N.; Ding, L.; Chen, H.; Wan, J.; Yang, X.; Xia, L.; He, W.; Xiong, H.; et al. The Interplay between the Gut Microbiota and NLRP3 Activation Affects the Severity of Acute Pancreatitis in Mice. Gut Microbes 2020, 11, 1774–1789. [Google Scholar] [CrossRef]
- Bell, J.K.; Siciliano, S.D.; Lamb, E.G. A Survey of Invasive Plants on Grassland Soil Microbial Communities and Ecosystem Services. Sci. Data 2020, 7, 86. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-In-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Goebel, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S RRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Catalogue of Life China: 2023 Annual Checklist. Available online: http://www.sp2000.org.cn (accessed on 24 August 2023).
- Borcard, D.; Gillet, F.; Legendre, P. Numerical Ecology with R; Springer: Cham, Switzerland, 2018; ISBN 9783319714035. [Google Scholar]
- Graf, D.; Monk, J.M.; Lepp, D.; Wu, W.; McGillis, L.; Roberton, K.; Brummer, Y.; Tosh, S.M.; Power, K.A. Cooked Red Lentils Dose-Dependently Modulate the Colonic Microenvironment in Healthy C57Bl/6 Male Mice. Nutrients 2019, 11, 1853. [Google Scholar] [CrossRef]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the Roles of Immigration and Chance in Shaping Prokaryote Community Structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Li, L.; Wu, C.; Huang, D.; Ding, C.; Wei, Y.; Sun, J. Integrating Stochastic and Deterministic Process in the Biogeography of N2-Fixing Cyanobacterium Candidatus Atelocyanobacterium Thalassa. Front. Microbiol. 2021, 12, 654646. [Google Scholar] [CrossRef]
- Hubbell, S.P. Luís Borda-de-Água The Unified Neutral Theory of Biodiversity and Biogeography. Ecology 2002, 83, 1771. [Google Scholar] [CrossRef]
- Chen, H.; Ma, Z. Niche-Neutral Continuum Seems to Explain the Global Niche Differentiation and Local Drift of the Human Digestive Tract Microbiome. Front. Microbiol. 2022, 13, 912240. [Google Scholar] [CrossRef] [PubMed]
- Laparra, J.M.; Sanz, Y. Interactions of Gut Microbiota with Functional Food Components and Nutraceuticals. Pharmacol. Res. 2010, 61, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Townsend, G.E.; Han, W.; Schwalm, N.D.; Hong, X.; Bencivenga-Barry, N.A.; Goodman, A.L.; Groisman, E.A. A Master Regulator of Bacteroides Thetaiotaomicron Gut Colonization Controls Carbohydrate Utilization and an Alternative Protein Synthesis Factor. mBio 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Baniel, A.; Amato, K.R.; Beehner, J.C.; Bergman, T.J.; Mercer, A.; Perlman, R.F.; Petrullo, L.; Reitsema, L.; Sams, S.; Lu, A.; et al. Seasonal Shifts in the Gut Microbiome Indicate Plastic Responses to Diet in Wild Geladas. Microbiome 2021, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.F.; Yang, Z.S.; Yao, R.; Xu, L.L.; Chen, H.; Gu, X.D.; Wu, T.G.; Yang, X.Y. Potential Mechanism of Detoxification of Cyanide Compounds by Gut Microbiomes of Bamboo-Eating Pandas. mSphere 2018, 3, 1–11. [Google Scholar] [CrossRef]
- Klure, D.M.; Dearing, M.D. Seasonal Restructuring Facilitates Compositional Convergence of Gut Microbiota in Free-Ranging Rodents. FEMS Microbiol. Ecol./FEMS Microbiol. Ecol. 2023, 99, fiad127. [Google Scholar] [CrossRef]
- Hu, X.L.; Liu, G.; Li, Y.M.; Wei, Y.T.; Lin, S.B.; Liu, S.Q.; Zheng, Y.L.; Hu, D.F. High-Throughput Analysis Reveals Seasonal Variation of the Gut Microbiota Composition within Forest Musk Deer (Moschus berezovskii). Front. Microbiol. 2018, 9, 1674. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.; Robson, P.; Farrar, K.; Thomas Jones, S.; Clifton-Brown, J.; Payne, R.; Donnison, I. TowardsMiscanthusCombustion Quality Improvement: The Role of Flowering and Senescence. GCB Bioenergy 2016, 9, 891–908. [Google Scholar] [CrossRef]
- John, D. Annual Lipid Cycles in Hibernators: Integration of Physiology and Behavior. Annu. Rev. Nutr. 2005, 25, 469–497. [Google Scholar] [CrossRef]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Müller, M.; de Vos, W.M. Modulation of Mucosal Immune Response, Tolerance, and Proliferation in Mice Colonized by the Mucin-Degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, S.; Wang, X.; Weng, Y.; Fan, X.; Sheng, H.; Zhu, X.; Lou, L.; Zhang, F. The Flavonoid-Rich Quzhou Fructus Aurantii Extract Modulates Gut Microbiota and Prevents Obesity in High-Fat Diet-Fed Mice. Nutr. Diabetes 2019, 9, 30. [Google Scholar] [CrossRef]
- Suzuki, T.A.; Martins, F.M.; Phifer-Rixey, M.; Nachman, M.W. The Gut Microbiota and Bergmann’s Rule in Wild House Mice. Mol. Ecol. 2020, 29, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.-C.; Lian, C.-A.; He, L.-S. Genomic Characterization of a Novel Tenericutes Bacterium from Deep-Sea Holothurian Intestine. Microorganisms 2020, 8, 1874. [Google Scholar] [CrossRef]
- Hullar, M.A.J.; Jenkins, I.C.; Randolph, T.W.; Curtis, K.R.; Monroe, K.R.; Ernst, T.; Shepherd, J.A.; Stram, D.O.; Cheng, I.; Kristal, B.S.; et al. Associations of the Gut Microbiome with Hepatic Adiposity in the Multiethnic Cohort Adiposity Phenotype Study. Gut Microbes 2021, 13, 1965463. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.E.; Kwon, M.-S.; Whon, T.W.; Kim, D.W.; Yun, M.; Lee, J.; Shin, M.-Y.; Kim, S.-H.; Choi, H.-J. Alteration of Gut Microbiota after Antibiotic Exposure in Finishing Swine. Front. Microbiol. 2021, 12, 596002. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yue, M.; Cheng, Y.; Sullivan, M.A.; Chen, W.; Yu, H.; Li, F.; Wu, S.; Lv, Y.; Zhai, X.; et al. Naringenin Prevents Non-Alcoholic Steatohepatitis by Modulating the Host Metabolome and Intestinal Microbiome in MCD Diet-Fed Mice. Food Sci. Nutr. 2023, 11, 7826–7840. [Google Scholar] [CrossRef]
- Combes, S.; Massip, K.; Martin, O.; Furbeyre, H.; Cauquil, L.; Pascal, G.; Bouchez, O.; Le Floc’h, N.; Zemb, O.; Oswald, I.P.; et al. Impact of Feed Restriction and Housing Hygiene Conditions on Specific and Inflammatory Immune Response, the Cecal Bacterial Community and the Survival of Young Rabbits. Animal 2017, 11, 854–863. [Google Scholar] [CrossRef]
- Berg, M.E.; Antonopoulos, D.A.; Rincón, M.T.; Band, M.; Bari, A.; Akraiko, T.V.; Hernández, Á.G.; Thimmapuram, J.; Henrissat, B.; Coutinho, P.M.; et al. Diversity and Strain Specificity of Plant Cell Wall Degrading Enzymes Revealed by the Draft Genome of Ruminococcus Flavefaciens FD-1. PLoS ONE 2009, 4, e6650. [Google Scholar] [CrossRef]
- López-Siles, M.; Khan, T.M.; Duncan, S.H.; Harmsen, M.; Garcia-Gil, L.; Garcia-Gil, H.J. Cultured Representatives of Two Major Phylogroups of Human Colonic Faecalibacterium prausnitzii Can Utilize Pectin, Uronic Acids, and Host-Derived Substrates for Growth. Appl. Environ. Microbiol. 2012, 78, 420–428. [Google Scholar] [CrossRef]
- Aarnoutse, R.; Ziemons, J.; Hillege, L.E.; de Vos-Geelen, J.; de Boer, M.; Bisschop, S.M.P.; Vriens, B.E.P.J.; Vincent, J.; van de Wouw, A.J.; Le, G.N.; et al. Changes in Intestinal Microbiota in Postmenopausal Oestrogen Receptor-Positive Breast Cancer Patients Treated with (Neo)Adjuvant Chemotherapy. NPJ Breast Cancer 2022, 8, 89. [Google Scholar] [CrossRef]
- Zhou, H.; Zeng, X.; Sun, D.; Chen, Z.; Chen, W.; Fan, L.; Limpanont, Y.; Dekumyoy, P.; Maleewong, W.; Lv, Z. Monosexual Cercariae of Schistosoma Japonicum Infection Protects against DSS-Induced Colitis by Shifting the Th1/Th2 Balance and Modulating the Gut Microbiota. Front. Microbiol. 2021, 11, 606605. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.C.; Healey, G.R.; Kelly, W.K.; Norris, G.E.; Jordens, Z.; Tannock, G.W.; Sims, I.R.; Bell, T.J.; Hedderley, D.; Henrissat, B.; et al. Genomic Insights from Monoglobus Pectinilyticus: A Pectin-Degrading Specialist Bacterium in the Human Colon. ISME J. 2019, 13, 1437–1456. [Google Scholar] [CrossRef]
- Haak, B.W.; Lankelma, J.M.; Hugenholtz, F.; Belzer, C.; de Vos, W.M.; Wiersinga, W.J. Long-Term Impact of Oral Vancomycin, Ciprofloxacin and Metronidazole on the Gut Microbiota in Healthy Humans. J. Antimicrob. Chemother. 2018, 74, 782–786. [Google Scholar] [CrossRef]
- Liu, C.; Finegold, S.M.; Song, Y.; Lawson, P.A. Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov. and Description of Blautia wexlerae sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2008, 58, 1896–1902. [Google Scholar] [CrossRef]
- Liu, X.; Mao, B.; Gu, J.; Wu, J.; Cui, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Blautia—A New Functional Genus with Potential Probiotic Properties? Gut Microbes 2021, 13, 1875796. [Google Scholar] [CrossRef]
- Jin, D.X.; Zou, H.W.; Liu, S.Q.; Wang, L.Z.; Xue, B.; Wu, D.; Tian, G.; Cai, J.; Yan, T.H.; Wang, Z.S.; et al. The Underlying Microbial Mechanism of Epizootic Rabbit Enteropathy Triggered by a Low Fiber Diet. Sci. Rep. 2018, 8, 12489. [Google Scholar] [CrossRef]
- Neumann, A.P.; Suen, G. The Phylogenomic Diversity of Herbivore-Associated Fibrobacter spp. Is Correlated to Lignocellulose-Degrading Potential. mSphere 2018, 3, 1–18. [Google Scholar] [CrossRef]
- Kohl, K.D.; Dieppa-Colón, E.; Goyco-Blas, J.; Peralta-Martínez, K.; Scafidi, L.; Shah, S.; Zawacki, E.; Barts, N.; Ahn, Y.; Hedayati, S.; et al. Gut Microbial Ecology of Five Species of Sympatric Desert Rodents in Relation to Herbivorous and Insectivorous Feeding Strategies. Integr. Comp. Biol. 2022, 62, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Schoener, T.W. Theory of Feeding Strategies. Annu. Rev. Ecol. Syst. 1971, 2, 369–404. [Google Scholar] [CrossRef]
- Trevail, A.M.; Green, J.A.; Sharples, J.; Polton, J.A.; Miller, P.I.; Daunt, F.; Owen, E.; Bolton, M.; Colhoun, K.; Newton, S.; et al. Environmental Heterogeneity Decreases Reproductive Success via Effects on Foraging Behaviour. Proc. R. Soc. B Biol. Sci. 2019, 286, 20190795. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial Degradation of Complex Carbohydrates in the Gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.K.; Williams, C.V.; Junge, R.E.; Mahefarisoa, K.L.; Rajaonarivelo, T.; Rakotondrainibe, H.; O’Connell, T.M.; Drea, C.M. A Role for Gut Microbiota in Host Niche Differentiation. ISME J. 2020, 14, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Carlos, C.; Fan, H.; Currie, C.R. Substrate Shift Reveals Roles for Members of Bacterial Consortia in Degradation of Plant Cell Wall Polymers. Front. Microbiol. 2018, 9, 364. [Google Scholar] [CrossRef]
- Xie, C.; Gong, W.; Zhu, Z.; Zhou, Y.; Xu, C.; Yan, L.; Hu, Z.; Ai, L.; Peng, Y. Comparative Secretome of White-Rot Fungi Reveals Co-Regulated Carbohydrate-Active Enzymes Associated with Selective Ligninolysis of Ramie Stalks. Microb. Biotechnol. 2020, 14, 911–922. [Google Scholar] [CrossRef]
- Borbón-García, A.; Reyes, A.; Vives-Flórez, M.J.; Caballero, S. Captivity Shapes the Gut Microbiota of Andean Bears: Insights into Health Surveillance. Front. Microbiol. 2017, 8, 1316. [Google Scholar] [CrossRef]
- Gudi, R.; Perez, N.; Johnson, B.M.; Sofi, M.H.; Brown, R.; Quan, S.; Karumuthil-Melethil, S.; Vasu, C. Complex Dietary Polysaccharide Modulates Gut Immune Function and Microbiota, and Promotes Protection from Autoimmune Diabetes. Immunology 2019, 157, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Douny, C.; Dufourny, S.; Brose, F.; Verachtert, P.; Rondia, P.; Lebrun, S.; Marzorati, M.; Everaert, N.; Delcenserie, V.; Scippo, M.L. Development of an Analytical Method to Detect Short-Chain Fatty Acids by SPME-GC–MS in Samples Coming from an in Vitro Gastrointestinal Model. J. Chromatogr. B 2019, 1124, 188–196. [Google Scholar] [CrossRef]
- Yu, H.R.; Sheen, J.M.; Hou, C.Y.; Lin, I.C.; Huang, L.T.; Tain, Y.L.; Cheng, H.H.; Lai, Y.J.; Lin, Y.J.; Tiao, M.M.; et al. Effects of Maternal Gut Microbiota-Targeted Therapy on the Programming of Nonalcoholic Fatty Liver Disease in Dams and Fetuses, Related to a Prenatal High-Fat Diet. Nutrients 2022, 14, 4004. [Google Scholar] [CrossRef]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative Analyses of Foregut and Hindgut Bacterial Communities in Hoatzins and Cows. ISME J. 2011, 6, 531–541. [Google Scholar] [CrossRef]
- Newsome, S.D.; Feeser, K.L.; Bradley, C.J.; Wolf, C.; Takacs-Vesbach, C.; Fogel, M.L. Isotopic and Genetic Methods Reveal the Role of the Gut Microbiome in Mammalian Host Essential Amino Acid Metabolism. Proc. R. Soc. B Biol. Sci. 2020, 287, 20192995. [Google Scholar] [CrossRef]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide Utilization by Gut Bacteria: Potential for New Insights from Genomic Analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Clauss, M.; Hume, I.D.; Hummel, J. Evolutionary Adaptations of Ruminants and Their Potential Relevance for Modern Production Systems. Animal 2010, 4, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Jewell, K.A.; McCormick, C.A.; Odt, C.L.; Weimer, P.J.; Suen, G. Ruminal Bacterial Community Composition in Dairy Cows Is Dynamic over the Course of Two Lactations and Correlates with Feed Efficiency. Appl. Environ. Microbiol. 2015, 81, 4697–4710. [Google Scholar] [CrossRef] [PubMed]
- Skarlupka, J.H.; Kamenetsky, M.E.; Jewell, K.A.; Suen, G. The Ruminal Bacterial Community in Lactating Dairy Cows Has Limited Variation on a Day-To-Day Basis. J. Anim. Sci. Biotechnol. 2019, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Shang, P.; Wei, M.; Duan, M.; Yan, F.; Chamba, Y. Healthy Gut Microbiome Composition Enhances Disease Resistance and Fat Deposition in Tibetan Pigs. Front. Microbiol. 2022, 13, 965292. [Google Scholar] [CrossRef] [PubMed]
- Déru, V.; Tiezzi, F.; Carillier-Jacquin, C.; Blanchet, B.; Cauquil, L.; Zemb, O.; Bouquet, A.; Maltecca, C.; Gilbert, H. Gut Microbiota and Host Genetics Contribute to the Phenotypic Variation of Digestive and Feed Efficiency Traits in Growing Pigs Fed a Conventional and a High Fiber Diet. Genet. Sel. Evol. 2022, 54, 55. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y. Cortical Microtubule Rearrangements and Cell Wall Patterning. Front. Plant Sci. 2015, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Patra, S.K.; Ghosh, S. Secretome Analysis of Macrophomina Phaseolina Identifies an Array of Putative Virulence Factors Responsible for Charcoal Rot Disease in Plants. Front. Microbiol. 2022, 13, 847832. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hameed, I.; Cao, D.; He, D.; Yang, P. Integrated Omics Analyses Identify Key Pathways Involved in Petiole Rigidity Formation in Sacred Lotus. Int. J. Mol. Sci. 2020, 21, 5087. [Google Scholar] [CrossRef]
- Beguin, P. Molecular Biology of Cellulose Degradation. Annu. Rev. Microbiol. 1990, 44, 219–248. [Google Scholar] [CrossRef]
- Bredon, M.; Dittmer, J.; Noël, C.; Moumen, B.; Bouchon, D. Lignocellulose Degradation at the Holobiont Level: Teamwork in a Keystone Soil Invertebrate. Microbiome 2018, 6, 162. [Google Scholar] [CrossRef]
- Suttner, B.; Johnston, E.R.; Orellana, L.H.; Rodriguez-R, L.M.; Hatt, J.K.; Carychao, D.; Carter, M.Q.; Cooley, M.B.; Konstantinidis, K.T. Metagenomics as a Public Health Risk Assessment Tool in a Study of Natural Creek Sediments Influenced by Agricultural and Livestock Runoff: Potential and Limitations. Appl. Environ. Microbiol. 2020, 86, e02525-19. [Google Scholar] [CrossRef] [PubMed]
- La Reau, A.J.; Suen, G. The Ruminococci: Key Symbionts of the Gut Ecosystem. J. Microbiol. 2018, 56, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hannula, S.E.; Van Veen, J.A. Primer Sets Developed for Functional Genes Reveal Shifts in Functionality of Fungal Community in Soils. Front. Microbiol. 2016, 7, 1897. [Google Scholar] [CrossRef]
- Berlemont, R.; Martiny, A.C. Phylogenetic Distribution of Potential Cellulases in Bacteria. Appl. Environ. Microbiol. 2012, 79, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Weimer, P.J. Cellulose Degradation by Ruminal Microorganisms. Crit. Rev. Biotechnol. 1992, 12, 189–223. [Google Scholar] [CrossRef]
- Podrepšek, G.H.; Knez, Ž.; Leitgeb, M. The Synthesis of (Magnetic) Crosslinked Enzyme Aggregates with Laccase, Cellulase, β-Galactosidase and Transglutaminase. Front. Bioeng. Biotechnol. 2022, 10, 813919. [Google Scholar] [CrossRef]
- Wang, G.; Yan, F.; Wang, Y.F.; Liu, Y.P.; Cui, J.N.; Yu, Z.L.; Feng, L.; James, T.D.; Wang, C.; Kong, Y. Visual Sensing of β-Glucosidase from Intestinal Fungus in the Generation of Cytotoxic Icarisid II. Front. Chem. 2022, 10, 919624. [Google Scholar] [CrossRef]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of Polyphenols on Gut Microbiota and Implications in Human Health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef]
- Kameshwar, A.K.S.; Qin, W. Metadata Analysis of Phanerochaete chrysosporium Gene Expression Data Identified Common CAZymes Encoding Gene Expression Profiles Involved in Cellulose and Hemicellulose Degradation. Int. J. Biol. Sci. 2017, 13, 85–99. [Google Scholar] [CrossRef]
- Gustaw, W.; Kordowska-Wiater, M.; Justyna, K. The Influence of Selected Prebiotics on the Growth of Lactic Acid Bacteria for Bio-Yoghurt Production. Acta Sci. Pol. Technol. Aliment. 2012, 10, 455–466. [Google Scholar]
- Mirande, C.; Kadlecikova, E.; Matulova, M.; Capek, P.; Bernalier-Donadille, A.; Forano, E.; Béra-Maillet, C. Dietary Fibre Degradation and Fermentation by Two Xylanolytic Bacteria Bacteroides Xylanisolvens XB1A and Roseburia Intestinalis XB6B4 from the Human Intestine. J. Appl. Microbiol. 2010, 109, 451–460. [Google Scholar] [CrossRef]
- Liao, Y.; Koelewijn, S.-F.; Van den Bossche, G.; Van Aelst, J.; Van den Bosch, S.; Renders, T.; Navare, K.; Nicolaï, T.; Van Aelst, K.; Maesen, M.; et al. A Sustainable Wood Biorefinery for Low–Carbon Footprint Chemicals Production. Science 2020, 367, 1385–1390. [Google Scholar] [CrossRef]
- Zhou, F.; Hansen, M.; Hobley, T.J.; Jensen, P.R. Valorization of Green Biomass: Alfalfa Pulp as a Substrate for Oyster Mushroom Cultivation. Foods 2022, 11, 2519. [Google Scholar] [CrossRef]
- Ravichandra, K.; Yaswanth, V.V.N.; Nikhila, B.; Ahmad, J.; Srinivasa Rao, P.; Uma, A.; Ravindrababu, V.; Prakasham, R.S. Xylanase Production by Isolated Fungal Strain, Aspergillus Fumigatus RSP-8 (MTCC 12039): Impact of Agro-Industrial Material as Substrate. Sugar Tech 2015, 18, 29–38. [Google Scholar] [CrossRef]
- Malherbe, S.; Cloete, T.E. Lignocellulose Biodegradation: Fundamentals and Applications. Rev. Environ. Sci. Bio/Technol. 2002, 1, 105–114. [Google Scholar] [CrossRef]
- Wang, Z.; Usyk, M.; Vázquez-Baeza, Y.; Chen, G.-C.; Isasi, C.R.; Williams-Nguyen, J.S.; Hua, S.; McDonald, D.; Thyagarajan, B.; Daviglus, M.L.; et al. Microbial Co-Occurrence Complicates Associations of Gut Microbiome with US Immigration, Dietary Intake and Obesity. Genome Biol. 2021, 22, 336. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, Z.; Chen, K.; Han, T.; Bu, F.; Sun, S.; Zhu, N.; Man, D.; Yang, K.; Yuan, S.; Fu, H. Natural Foraging Selection and Gut Microecology of Two Subterranean Rodents from the Eurasian Steppe in China. Animals 2024, 14, 2334. https://doi.org/10.3390/ani14162334
Shang Z, Chen K, Han T, Bu F, Sun S, Zhu N, Man D, Yang K, Yuan S, Fu H. Natural Foraging Selection and Gut Microecology of Two Subterranean Rodents from the Eurasian Steppe in China. Animals. 2024; 14(16):2334. https://doi.org/10.3390/ani14162334
Chicago/Turabian StyleShang, Zhenghaoni, Kai Chen, Tingting Han, Fan Bu, Shanshan Sun, Na Zhu, Duhu Man, Ke Yang, Shuai Yuan, and Heping Fu. 2024. "Natural Foraging Selection and Gut Microecology of Two Subterranean Rodents from the Eurasian Steppe in China" Animals 14, no. 16: 2334. https://doi.org/10.3390/ani14162334
APA StyleShang, Z., Chen, K., Han, T., Bu, F., Sun, S., Zhu, N., Man, D., Yang, K., Yuan, S., & Fu, H. (2024). Natural Foraging Selection and Gut Microecology of Two Subterranean Rodents from the Eurasian Steppe in China. Animals, 14(16), 2334. https://doi.org/10.3390/ani14162334