Screening for Genes Related to Meat Production Traits in Duroc × Bama Xiang Crossbred Pigs by Whole Transcriptome Sequencing

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Sample Preparation
2.2. Determination of Carcass and Meat Phenotypic Traits and Plasma Biochemical Indicators
2.3. NADH-TR Staining and HE Staining of Myofibers
2.4. Library Preparation and RNA Sequencing
2.5. Differential Expression and Functional Analysis of mRNA, miRNA, and lncRNA
2.6. Quantitative Real-Time PCR (qRT-PCR) Validation
2.7. Analysis of ceRNA Regulatory Network
2.8. Statistical Analysis
3. Results
3.1. Analysis of Carcass and Meat Phenotypic Traits and Plasma Biochemical Indicators
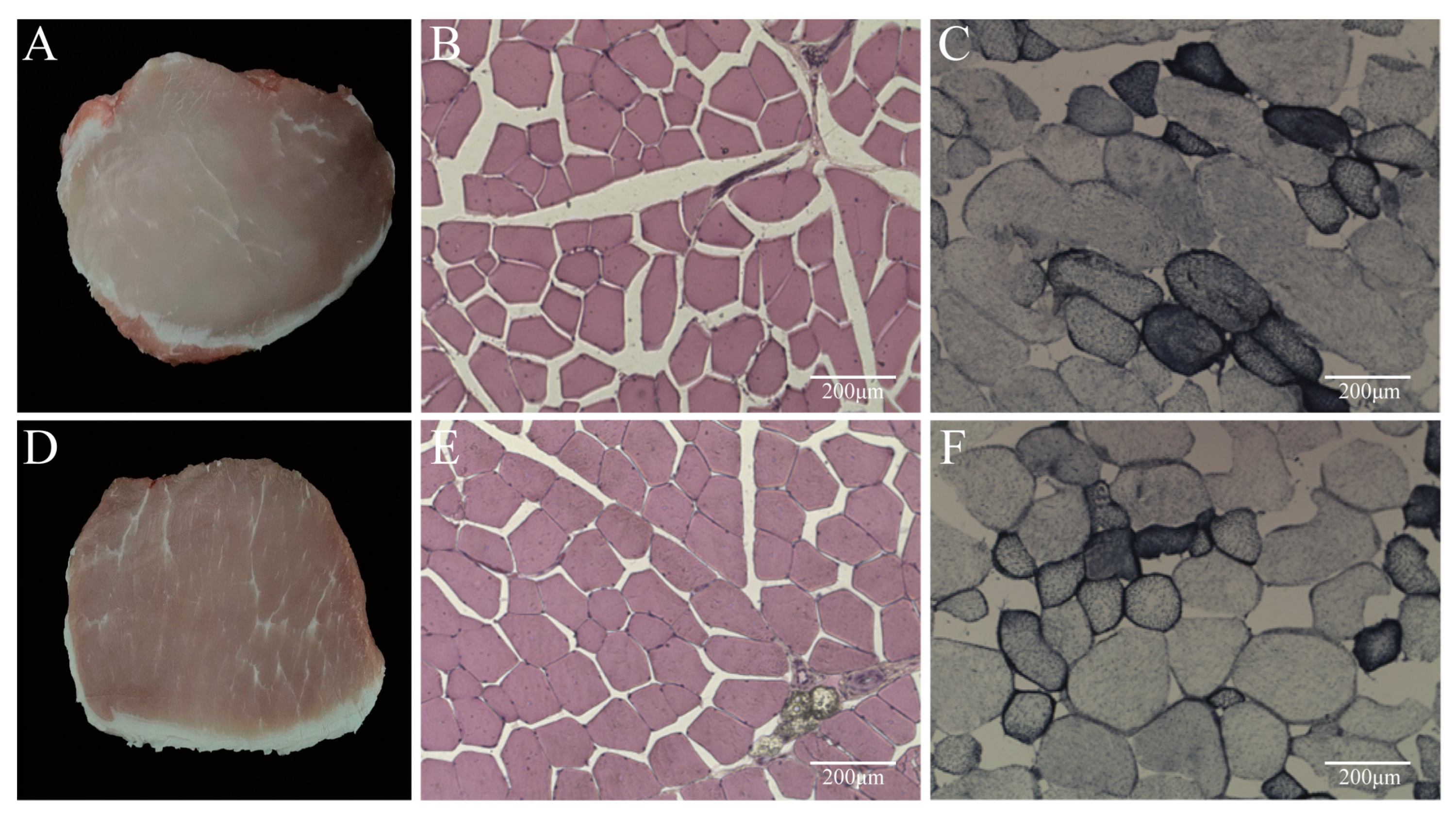
3.2. Comparison of Muscle Histological Properties and Muscle Fiber Types
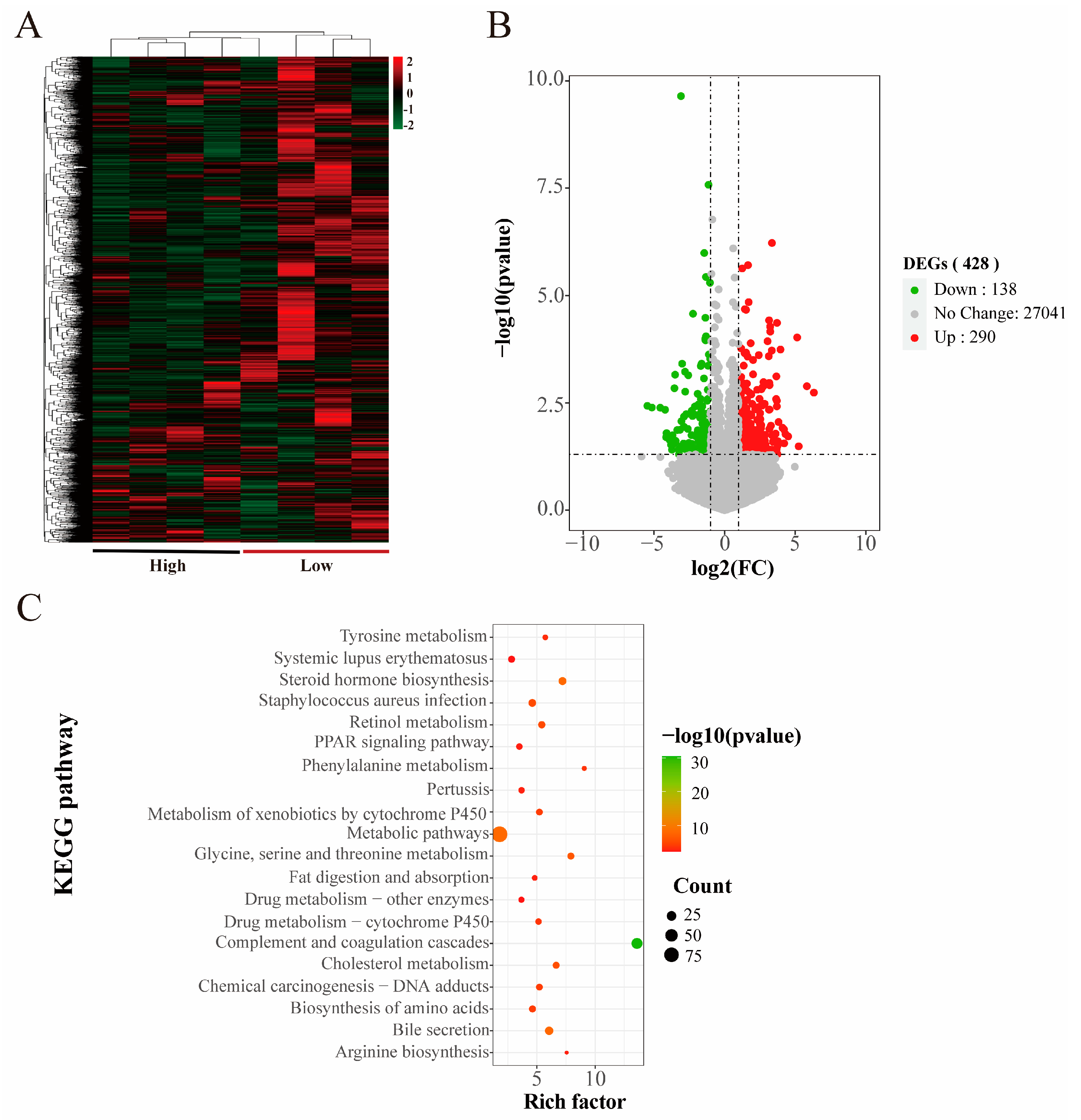
3.3. mRNA Sequencing Expression Analysis
3.4. miRNA Sequencing Expression Analysis
3.5. lncRNA Sequencing Expression Analysis
3.6. ceRNA Regulatory Network Analysis and Quantitative Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Albuquerque, A.; Óvilo, C.; Núñez, Y.; Benítez, R.; López-Garcia, A.; García, F.; Félix, M.d.R.; Laranjo, M.; Charneca, R.; Martins, J.M. Comparative Transcriptomic Analysis of Subcutaneous Adipose Tissue from Local Pig Breeds. Genes 2020, 11, 422. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.D.; Enser, M.; Fisher, A.V.; Nute, G.R.; Sheard, P.R.; Richardson, R.I.; Hughes, S.I.; Whittington, F.M. Fat deposition, fatty acid composition and meat quality: A review. Meat Sci. 2007, 78, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Prather, R.S. Pig genomics for biomedicine. Nat. Biotechnol. 2013, 31, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, J.; Xu, C.; Ma, N.; He, T.; Zhao, J.; Ma, X.; Thacker, P.A. Progress towards pig nutrition in the last 27 years. Sci. Food Agric. 2018, 100, 5102–5110. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhao, P.; Zheng, X.; Zhou, L.; Wang, C.; Liu, J.F. Genome-Wide Detection of Selection Signatures in Duroc Revealed Candidate Genes Relating to Growth and Meat Quality. G3 (Bethesda Md.) 2020, 10, 3765–3773. [Google Scholar] [CrossRef]
- Jiao, Y.; Hao, L.; Xia, P.; Cheng, Y.; Song, J.; Chen, X.; Wang, Z.; Ma, Z.; Zheng, S.; Chen, T.; et al. Identification of Potential miRNA–mRNA Regulatory Network Associated with Pig Growth Performance in the Pituitaries of Bama Minipigs and Landrace Pigs. Animals 2022, 12, 3058. [Google Scholar] [CrossRef]
- Mo, J.; Lu, Y.; Zhu, S.; Feng, L.; Qi, W.; Chen, X.; Xie, B.; Chen, B.; Lan, G.; Liang, J. Genome-Wide Association Studies, Runs of Homozygosity Analysis, and Copy Number Variation Detection to Identify Reproduction-Related Genes in Bama Xiang Pigs. Front. Vet. Sci. 2022, 9, 892815. [Google Scholar] [CrossRef]
- Liu, Y.; He, Q.; Azad, M.A.; Xiao, Y.; Yin, Y.; Kong, X. Nuclear Magnetic Resonance-Based Metabolomic Analysis Reveals Physiological Stage, Breed, and Diet Effects on the Intramuscular Metabolism of Amino Acids and Related Nutrients in Pigs. Front. Vet. Sci. 2021, 8, 681192. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yu, Z.; Ai, N.; Liufu, S.; Liu, X.; Chen, B.; Li, X.; Jiang, J.; Zhang, Y.; Ma, H.; et al. Molecular Mechanism of MYL4 Regulation of Skeletal Muscle Development in Pigs. Genes 2023, 14, 1267. [Google Scholar] [CrossRef]
- Tang, Z.; Li, Y.; Wan, P.; Li, X.; Zhao, S.; Liu, B.; Fan, B.; Zhu, M.; Yu, M.; Li, K. LongSAGE analysis of skeletal muscle at three prenatal stages in Tongcheng and Landrace pigs. Genome Biol. 2007, 8, R115. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Li, J.; Li, S.; Cao, H.; Jin, X.; Zeng, Y.; Chen, W. Full-Length Transcriptome Analysis of Skeletal Muscle of Jiangquan Black Pig at Different Developmental Stages. Int. J. Mol. Sci. 2024, 25, 6095. [Google Scholar] [CrossRef] [PubMed]
- Hoa, V.B.; Seong, P.N.; Cho, S.H.; Kang, S.M.; Kim, Y.S.; Moon, S.S.; Choi, Y.M.; Kim, J.H.; Seol, K.H. Quality characteristics and flavor compounds of pork meat as a function of carcass quality grade. Asian-Australas. J. Anim. Sci. 2019, 32, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, S.; Zhang, X.; Ren, S.; Tang, Z.; Gao, F. Coordinated transcriptional and post-transcriptional epigenetic regulation during skeletal muscle development and growth in pigs. J. Anim. Sci. Biotechnol. 2022, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Costa, V.; Angelini, C.; De Feis, I.; Ciccodicola, A. Uncovering the Complexity of Transcriptomes with RNA-Seq. J. Biomed. Biotechnol. 2010, 2010, 853916. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, X.; Li, R.; Michal, J.J.; Zhang, S.; Dodson, M.V.; Zhang, Z.; Harland, R.M. Whole transcriptome analysis with sequencing: Methods, challenges and potential solutions. Cell. Mol. Life Sci. CMLS 2015, 72, 3425–3439. [Google Scholar] [CrossRef]
- Gan, L.; Xie, L.; Zuo, F.; Xiang, Z.; He, N. Transcriptomic analysis of Rongchang pig brains and livers. Genes 2015, 560, 96–106. [Google Scholar] [CrossRef]
- Ayuso, M.; Fernández, A.; Núñez, Y.; Benítez, R.; Isabel, B.; Barragán, C.; Fernández, A.I.; Rey, A.I.; Medrano, J.F.; Cánovas, Á.; et al. Comparative Analysis of Muscle Transcriptome between Pig Genotypes Identifies Genes and Regulatory Mechanisms Associated to Growth, Fatness and Metabolism. PLoS ONE 2017, 10, e0145162. [Google Scholar] [CrossRef]
- Ghosh, M.; Sodhi, S.S.; Song, K.D.; Kim, J.H.; Mongre, R.K.; Sharma, N.; Singh, N.K.; Kim, S.W.; Lee, H.K.; Jeong, D.K. Evaluation of body growth and immunity-related differentially expressed genes through deep RNA sequencing in the piglets of Jeju native pig and Berkshire. Anim. Genet. 2015, 46, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Te Pas, M.F.; De Wit, A.A.; Priem, J.; Cagnazzo, M.; Davoli, R.; Russo, V.; Pool, M.H. Transcriptome expression profiles in prenatal pigs in relation to myogenesis. J. Muscle Res. Cell Motil. 2005, 26, 157–165. [Google Scholar] [CrossRef]
- Xing, K.; Zhao, X.; Ao, H.; Chen, S.; Yang, T.; Tan, Z.; Wang, Y.; Zhang, F.; Liu, Y.; Ni, H.; et al. Transcriptome analysis of miRNA and mRNA in the livers of pigs with highly diverged backfat thickness. Sci. Rep. 2019, 9, 16740. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef]
- Xu, Y.; Qi, X.; Hu, M.; Lin, R.; Hou, Y.; Wang, Z.; Zhou, H.; Zhao, Y.; Luan, Y.; Zhao, S.; et al. Transcriptome Analysis of Adipose Tissue Indicates That the cAMP Signaling Pathway Affects the Feed Efficiency of Pigs. Genes 2018, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, S.; Yuan, J.; Shi, Y.; Wang, Y.; Xi, Y.; Qi, X.; Guo, Y.; Sheng, X.; Liu, J.; et al. Comprehensive Analysis of the lncRNA–miRNA–mRNA Regulatory Network for Intramuscular Fat in Pigs. Genes 2023, 14, 168. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tian, X.; Li, D.; He, Y.; Yang, P.; Cheng, Y.; Zhao, X.; Sun, J.; Yang, G. Transcriptome, proteome and metabolome analysis provide insights on fat deposition and meat quality in pig. Food Res. Int. 2023, 166, 112550. [Google Scholar] [CrossRef]
- Zhang, J.; Chai, J.; Luo, Z.; He, H.; Chen, L.; Liu, X.; Zhou, Q. Meat and nutritional quality comparison of purebred and crossbred pigs. Anim. Sci. J. Nihon Chikusan Gakkaiho 2018, 89, 202–210. [Google Scholar] [CrossRef]
- Madeira, M.S.; Lopes, P.A.; Costa, P.; Coelho, D.; Alfaia, C.M.; Prates, J.A.M. Reduced protein diets increase intramuscular fat of psoas major, a red muscle, in lean and fatty pig genotypes. Animals 2017, 11, 2094–2102. [Google Scholar] [CrossRef]
- Chen, C.; Zheng, J.; Xiong, C.; Zhou, H.; Wei, C.; Hu, X.; Qian, X.; He, M.; Shi, Y.; Liu, Y.; et al. Metabolomics Characterize the Differential Metabolic Markers between Bama Xiang Pig and Debao Pig to Identify Pork. Foods 2022, 12, 5. [Google Scholar] [CrossRef]
- Li, X.; Lu, L.; Tong, X.; Li, R.; Jin, E.; Ren, M.; Gao, Y.; Gu, Y.; Li, S. Transcriptomic Profiling of Meat Quality Traits of Skeletal Muscles of the Chinese Indigenous Huai Pig and Duroc Pig. Genes 2023, 14, 1548. [Google Scholar] [CrossRef]
- Gozalo-Marcilla, M.; Buntjer, J.; Johnsson, M.; Batista, L.; Diez, F.; Werner, C.R.; Chen, C.Y.; Gorjanc, G.; Mellanby, R.J.; Hickey, J.M.; et al. Genetic architecture and major genes for backfat thickness in pig lines of diverse genetic backgrounds. Genet. Sel. Evol. 2021, 53, 76. [Google Scholar] [CrossRef]
- Nakajima, I.; Kojima, M.; Oe, M.; Ojima, K.; Muroya, S.; Chikuni, K. Comparing pig breeds with genetically low and high backfat thickness: Differences in expression of adiponectin, its receptor, and blood metabolites. Domest. Anim. Endocrinol. 2019, 68, 54–63. [Google Scholar] [CrossRef]
- Van Laack, R.; Kauffman, R.; Greaser, M. Determinants of ultimate pH of meat. In Proceedings of the International Congress of Meat Science and Technology, Krakow, Poland, 26–31 August 2001; pp. 22–27. [Google Scholar]
- Pipek, P.; Haberl, A.; Jelenikova, J. Influence of slaughterhouse handling on the quality of beef carcasses. Czech J. Anim. Sci. 2003, 48, 371–378. [Google Scholar]
- Villarroel, M.; María, G.A.; Sañudo, C.; Olleta, J.L.; Gebresenbet, G. Effect of transport time on sensorial aspects of beef meat quality. Meat Sci. 2003, 63, 353–357. [Google Scholar] [CrossRef]
- Won, S.; Jung, J.; Park, E.; Kim, H. Identification of genes related to intramuscular fat content of pigs using genome-wide association study. Asian-Australas. J. Anim. Sci. 2018, 31, 157–162. [Google Scholar] [CrossRef]
- Verbeke, W.V.; Van Oeckel, M.J.; Warnants, N.; Viaene, J.; Boucque, C.V. Consumer perception, facts and possibilities to improve acceptability of health and sensory characteristics of pork. Meat Sci. 1999, 53, 77–99. [Google Scholar] [CrossRef]
- Wood, R.J. Vitamin D and adipogenesis: New molecular insights. Nutr. Rev. 2008, 66, 40–46. [Google Scholar] [CrossRef]
- Muñoz, M.; García-Casco, J.M.; Caraballo, C.; Fernández-Barroso, M.Á.; Sánchez-Esquiliche, F.; Gómez, F.; Rodríguez, M.D.; Silió, L. Identification of Candidate Genes and Regulatory Factors Underlying Intramuscular Fat Content Through Longissimus Dorsi Transcriptome Analyses in Heavy Iberian Pigs. Front. Genet. 2018, 9, 608. [Google Scholar] [CrossRef]
- Anderson, J.W. Dietary fiber, lipids and atherosclerosis. Am. J. Cardiol. 1987, 60, G17–G22. [Google Scholar] [CrossRef]
- Huang, C.; Zhong, L.; Zou, X.; Huang, Y.; Cai, L.; Ma, J. Evidence Against the Causal Relationship Between a Putative Cis-Regulatory Variant of MYH3 and Intramuscular Fat Content in Pigs. Front. Vet. Sci. 2021, 8, 672852. [Google Scholar] [CrossRef]
- Wang, T.; Xu, Y.Q.; Yuan, Y.X.; Xu, P.W.; Zhang, C.; Li, F.; Wang, L.N.; Yin, C.; Zhang, L.; Cai, X.C.; et al. Succinate induces skeletal muscle fiber remodeling via SUCNR1 signaling. EMBO Rep. 2021, 22, e53027. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, S.; Kim, J.M. Genetic correlation between biopsied and post-mortem muscle fibre characteristics and meat quality traits in swine. Meat Sci. 2022, 186, 108735. [Google Scholar] [CrossRef] [PubMed]
- Bérard, J.; Kalbe, C.; Lösel, D.; Tuchscherer, A.; Rehfeldt, C. Potential sources of early-postnatal increase in myofibre number in pig skeletal muscle. Histochem. Cell Biol. 2011, 136, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Vrecl, M.; Cotman, M.; Uršič, M.; Čandek-Potokar, M.; Fazarinc, G. Age-Dependent Expression of MyHC Isoforms and Lipid Metabolism-Related Genes in the Longissimus Dorsi Muscle of Wild and Domestic Pigs. Animals 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, P. Neuromuscular Aspects of Physical Activity; Human Kinetics: Champaign, IL, USA, 2001; p. 256. [Google Scholar]
- Huang, Y.N.; Ao, Q.W.; Jiang, Q.Y.; Guo, Y.F.; Lan, G.Q.; Jiang, H.S. Comparisons of different myosin heavy chain types, AMPK, and PGC-1α gene expression in the longissimus dorsi muscles in Bama Xiang and Landrace pigs. Genet. Mol. Res. GMR 2016, 15, 10–4238. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Moon, S.S.; Song, S.; Cheng, H.; Im, C.; Du, L.; Kim, G.D. Comparative review of muscle fiber characteristics between porcine skeletal muscles. J. Anim. Sci. Technol. 2024, 66, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Bryson-Richardson, R.J.; Currie, P.D. The genetics of vertebrate myogenesis. Nature reviews. Genetics 2008, 9, 632–646. [Google Scholar]
- Li, X.J.; Zhou, J.; Liu, L.Q.; Qian, K.; Wang, C.L. Identification of genes in longissimus dorsi muscle differentially expressed between Wannanhua and Yorkshire pigs using RNA-sequencing. Anim. Genet. 2016, 47, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xi, Y.; Liu, G.; Zhao, Y.; Li, J.; Lei, M. Comparative transcriptomic analysis of skeletal muscle tissue during prenatal stages in Tongcheng and Yorkshire pig using RNA-seq. Funct. Integr. Genom. 2018, 18, 195–209. [Google Scholar] [CrossRef]
- Xu, J.; Wang, C.; Jin, E.; Gu, Y.; Li, S.; Li, Q. Identification of differentially expressed genes in longissimus dorsi muscle between Wei and Yorkshire pigs using RNA sequencing. Genes Genom. 2018, 40, 413–421. [Google Scholar] [CrossRef]
- Zeng, Q.; Du, Z. Advances in the discovery of genetic elements underlying longissimus dorsi muscle growth and development in the pig. Anim. Genet. 2023, 54, 709–720. [Google Scholar] [CrossRef]
- O’Donohue, T.; Sait, S.F.; Bender, J.G. Progress in precision therapy in pediatric oncology. Curr. Opin. Pediatrics. 2022, 35, 41–47. [Google Scholar] [CrossRef]
- Fan, H.; Ge, Y.; Ma, X.; Li, Z.; Shi, L.; Lin, L.; Xiao, J.; Chen, W.; Ni, P.; Yang, L.; et al. Long non-coding RNA CCDC144NL-AS1 sponges miR-143-3p and regulates MAP3K7 by acting as a competing endogenous RNA in gastric cancer. Cell Death Dis. 2020, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Chun, J.; Jung, H.A.; Choi, J.S.; Kim, Y.S. Capillarisin attenuates exercise-induced muscle damage through MAPK and NF-κB signaling. Phytomedicine 2017, 32, 30–36. [Google Scholar] [CrossRef]
- Tadashi, Y.; Patrice, D. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Kaya-Copur, A.; Marchiano, F.; Hein, M.Y.; Alpern, D.; Russeil, J.; Luis, N.M.; Mann, M.; Deplancke, B.; Habermann, B.H.; Schnorrer, F. The Hippo pathway controls myofibril assembly and muscle fiber growth by regulating sarcomeric gene expression. Elife 2021, 10, e63726. [Google Scholar] [CrossRef]
- Giuseppe, S.; Anna, P.; Riccardo, C.; José, C.H.; Emanuele, M. Mammalian Target of Rapamycin (mTOR) Signaling at the Crossroad of Muscle Fiber Fate in Sarcopenia. Int. J. Mol. Sci. 2022, 23, 13823. [Google Scholar] [CrossRef]
- Ravikumar, M.; Kalina, D.; Walter, W. PPARs and Microbiota in Skeletal Muscle Health and Wasting. Int. J. Mol. Sci. 2020, 21, 8056. [Google Scholar] [CrossRef]
- Gu, H.; Zhou, Y.; Yang, J.; Li, J.; Peng, Y.; Zhang, X.; Miao, Y.; Jiang, W.; Bu, G.; Hou, L.; et al. Targeted overexpression of PPARγ in skeletal muscle by random insertion and CRISPR/Cas9 transgenic pig cloning enhances oxidative fiber formation and intramuscular fat deposition. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21308. [Google Scholar] [CrossRef]
- Wen, N.; Yu, M.F.; Liu, J.; Cai, C.; Liu, Q.H.; Shen, J. Deficiency of MTMR14 impairs male fertility in Mus musculus. PLoS ONE 2018, 13, e0206224. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, M.S.; Chen, Y.; Liu, R.; Tan, X.; Li, W.; El-Wardany, I.; Zhao, D.; Zheng, M.; Wen, J.; Zhao, G. Paternal Dietary Methionine Supplementation Improves Carcass Traits and Meat Quality of Chicken Progeny. Animals 2021, 11, 325. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Wan, X.; Wu, R.; Yu, X.; Li, H.; Zhong, R.; Zhu, D.; Zhang, Y. Linc-RAM promotes muscle cell differentiation via regulating glycogen phosphorylase activity. J. Cell Regen. 2022, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; He, J.; Wei, W.; Chu, W.; Yu, S.; Tian, Y.; Peng, F.; Liu, H.; Zhang, Z.; Chen, J. The c.–360 T> C mutation affects PGAM2 transcription activity and is linked with the water holding capacity of the longissimus lumborum muscle in pigs. Meat Sci. 2016, 122, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bi, H.; Xie, S.; Cui, W. MiR-208b Regulates the Conversion of Skeletal Muscle Fiber Types by Inhibiting Mettl8 Expression. Front. Genet. 2022, 13, 820464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, Y.; Wang, L.; Liu, X.; Yan, H.; Gao, H.; Hou, X.; Zhang, Y.; Guo, H.; Yue, J.; et al. Genomic variants associated with the number and diameter of muscle fibers in pigs as revealed by a genome-wide association study. Animals 2019, 14, 475–481. [Google Scholar] [CrossRef]
- Esteve, A.; Ojeda, A.; Huang, L.S.; Folch, J.M.; Pérez-Enciso, M. Nucleotide variability of the porcine SERPINA6 gene and the origin of a putative causal mutation associated with meat quality. Anim. Genet. 2011, 42, 235–241. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurement Index | High Group 1 | Low Group 1 | |
|---|---|---|---|
| Live Weight at Slaughter (kg) | 148.38 ± 8.97 A | 88.75 ± 9.13 B | |
| Percentage of Skin Fat (%) | 40.74 ± 6.02 | 40.73 ± 2.80 | |
| Percentage of Lean Meat (%) | 44.05 ± 3.30 | 43.17 ± 2.81 | |
| Dressing Percentage (%) | 76.76 ± 1.09 | 78.08 ± 3.46 | |
| Loin Eye Area (cm2) | 36.13 ± 6.07 a | 25.72 ± 4.90 b | |
| Backfat Thickness (mm) | Middle Shoulder | 57.83 ± 12.87 | 58.83 ± 8.73 |
| Thoracolumbar Junction | 29.27 ± 8.12 b | 35.58 ± 4.54 a | |
| Lumbar Sacral Vertebra Junction | 31.47 ± 11.07 | 32.32 ± 2.57 | |
| pH 45 min | 6.51 ± 0.10 | 6.40 ± 0.24 | |
| pH 24 h | 5.69 ± 0.04 a | 5.60 ± 0.14 b | |
| Meat Color 45 min | 85.43 ± 2.45 | 84.85 ± 2.55 | |
| Meat Color 24 h | 65.58 ± 5.30 | 63.03 ± 6.38 | |
| Colorimetric Meat Color Score 45 min | 3.50 ± 0.58 | 3.00 ± 0.00 | |
| Colorimetric Meat Color Score 24 h | 2.00 ± 0.00 | 2.00 ± 0.00 | |
| Drip Loss 48 h (g) | 0.54 ± 0.17 | 0.48 ± 0.19 | |
| Share Force (KGF) | 7.82 ± 0.82 | 7.95 ± 1.52 | |
| Moisture (%) | 74.17 ± 0.01 | 73.49 ± 0.01 | |
| IMF (%) | 2.60 ± 0.008 b | 3.99 ± 0.015 a | |
| Plasma Metabolites | High Group 1 | Low Group 1 |
|---|---|---|
| Total Protein (mg/mL) | 31.20 ± 3.00 b | 36.23 ± 0.40 a |
| Cholesterol (mmol/mL) | 4.03 ± 0.47 b | 5.67 ± 0.92 a |
| Triglycerides (mmol/mL) | 0.86 ± 0.38 | 1.21 ± 0.19 |
| Creatinine (umol/mL) | 630.39 ± 69.83 | 534.91 ± 72.98 |
| Glucose (mg/mL) | 1.82 ± 0.17 | 1.74 ± 0.33 |
| Urea Nitrogen (mmol/mL) | 5.03 ± 1.60 | 7.40 ± 0.55 |
| Items | High Group 1 | Low Group 1 |
|---|---|---|
| Muscle Fiber Diameter (μm) | 40.87 ± 3.85 | 40.34 ± 5.68 |
| Myofiber Type | High Group 1 | Low Group 1 |
|---|---|---|
| Type I Muscle Fibers (%) | 14.1 ± 1.61 b | 19.9 ± 1.18 a |
| Type II Muscle Fibers (%) | 85.9 ± 4.77 a | 80.1 ± 5.30 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wang, H.; Cheng, F.; Chen, K.; Lei, G.; Deng, Z.; Wu, X.; Liu, C.; Si, J.; Liang, J. Screening for Genes Related to Meat Production Traits in Duroc × Bama Xiang Crossbred Pigs by Whole Transcriptome Sequencing. Animals 2024, 14, 2347. https://doi.org/10.3390/ani14162347
Xu Y, Wang H, Cheng F, Chen K, Lei G, Deng Z, Wu X, Liu C, Si J, Liang J. Screening for Genes Related to Meat Production Traits in Duroc × Bama Xiang Crossbred Pigs by Whole Transcriptome Sequencing. Animals. 2024; 14(16):2347. https://doi.org/10.3390/ani14162347
Chicago/Turabian StyleXu, Yupei, Hui Wang, Feng Cheng, Kuirong Chen, Guofeng Lei, Zhongrong Deng, Xiaoxiao Wu, Cong Liu, Jinglei Si, and Jing Liang. 2024. "Screening for Genes Related to Meat Production Traits in Duroc × Bama Xiang Crossbred Pigs by Whole Transcriptome Sequencing" Animals 14, no. 16: 2347. https://doi.org/10.3390/ani14162347