
Genome-Wide Association Analysis Identifies Important Haplotypes and Candidate Gene XKR4 for Body Size Traits in Pekin Ducks

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotypes and Genotypes
2.2. Statistical Analysis
2.3. Post GWAS
3. Results and Discussion
3.1. Phenotypes and Heritability
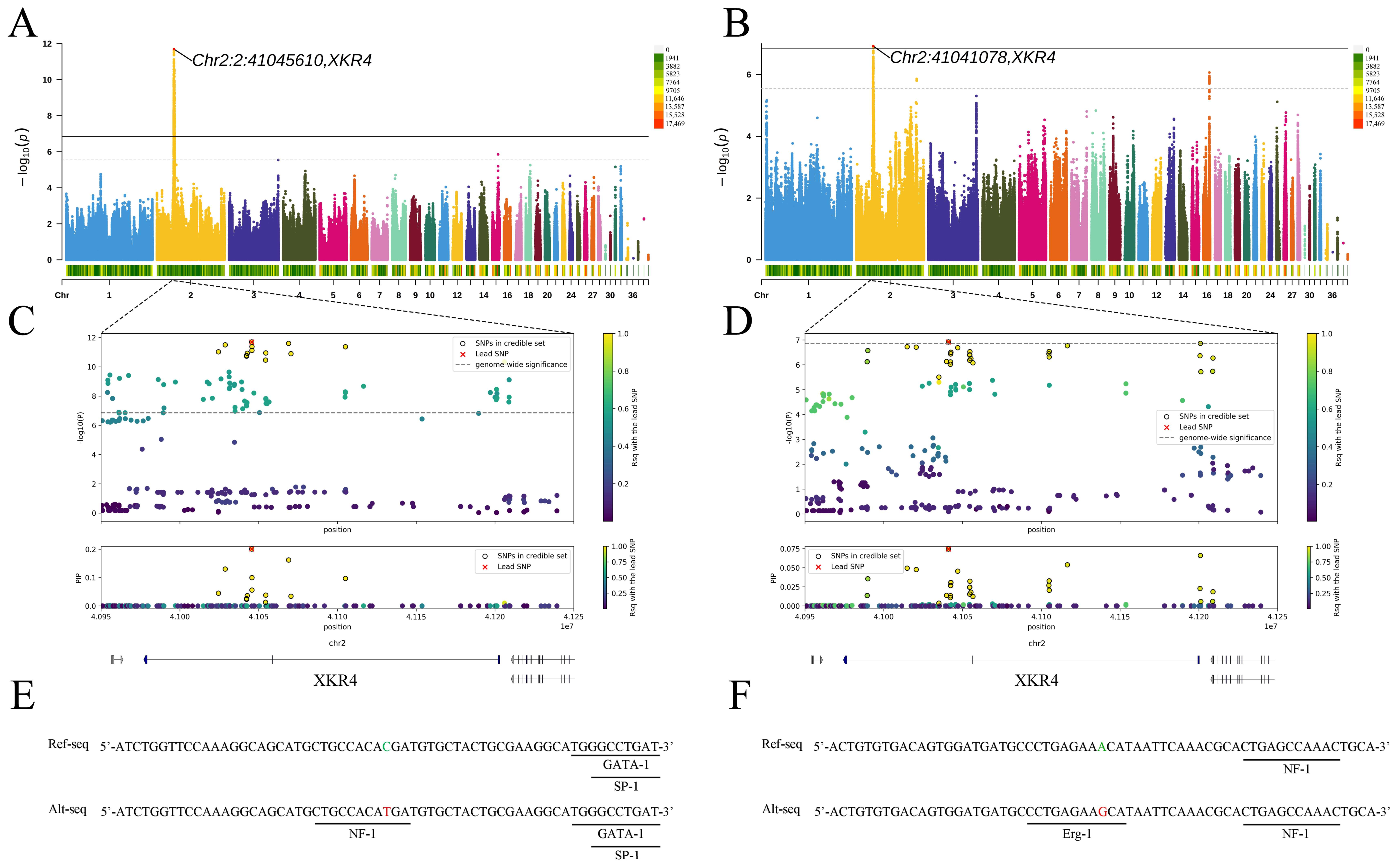
3.2. GWAS and TFBS Prediction
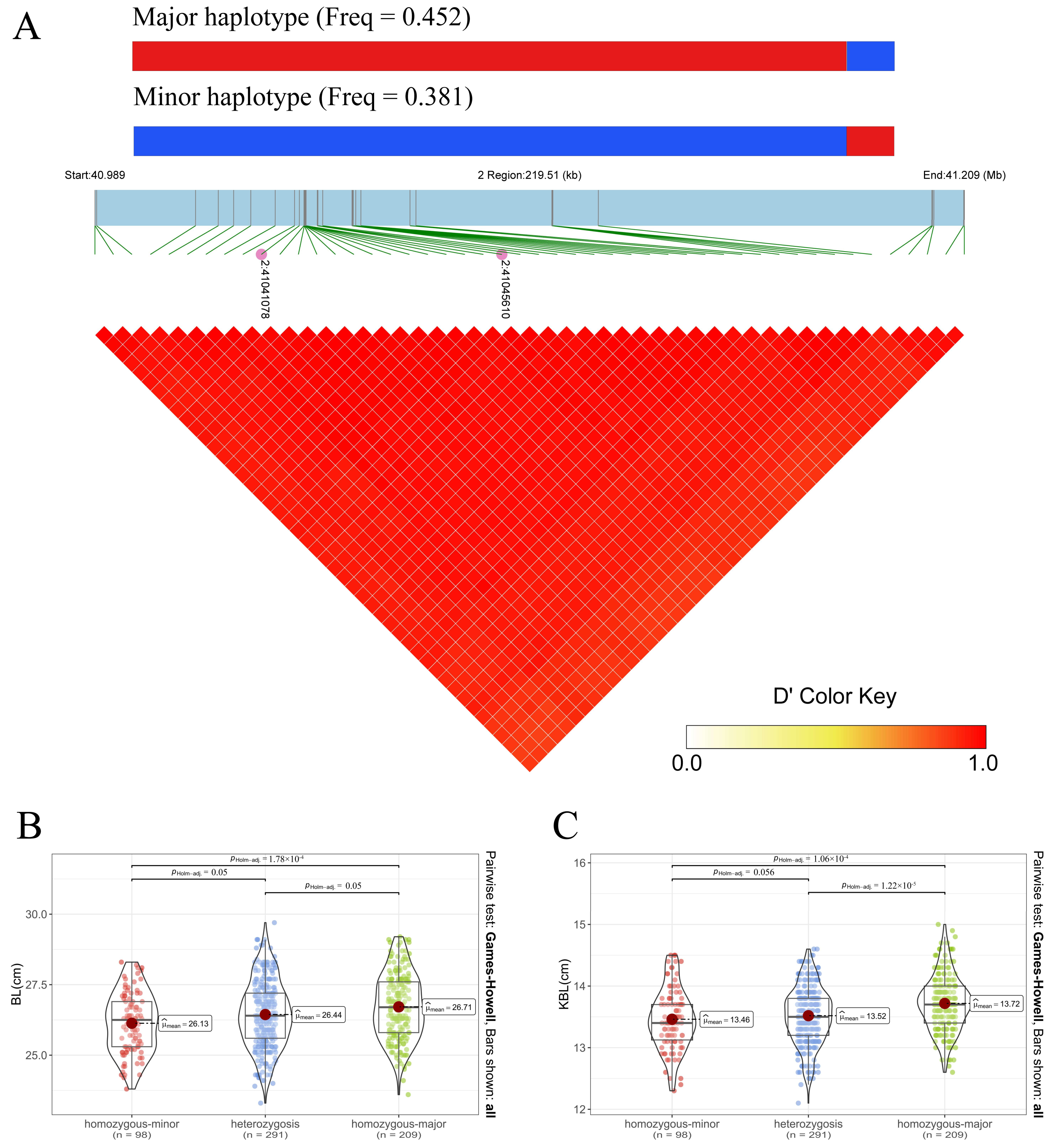
3.3. Fine-Mapping and Haplotype Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jalaludeen, A.; Churchil, R.; Baeza, E. Duck Production and Management Strategies; Springer: Singapore, 2022. [Google Scholar]
- Lyu, S.; Arends, D.; Nassar, M.K.; Weigend, A.; Weigend, S.; Wang, E.; Brockmann, G.A. High-density genotyping reveals candidate genomic regions for chicken body size in breeds of Asian origin. Poult. Sci. 2023, 102, 102303. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, C.G.; Song, C.; Du, Z.Q.; Deng, X.M.; Li, N.; Hu, X.X. Mapping quantitative trait loci affecting chicken body size traits via genome scanning. Anim. Genet. 2011, 42, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Dong, X.; Mao, H.; Xu, N.; Yin, Z. Expression Analysis of the PITX2 Gene and Associations between Its Polymorphisms and Body Size and Carcass Traits in Chickens. Animals 2019, 9, 1001. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Ye, J.; Dong, L.; Li, Y.; Yan, L.; Cai, G.; Liu, D.; Tan, C.; Wu, Z. Genome-Wide Association Study for Body Length, Body Height, and Total Teat Number in Large White Pigs. Front. Genet. 2021, 12, 650370. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, M.; Cheng, H.; Fan, W.; Yuan, Z.; Gao, Q.; Xu, Y.; Guo, Z.; Zhang, Y.; Hu, J.; et al. An intercross population study reveals genes associated with body size and plumage color in ducks. Nat. Commun. 2018, 9, 2648. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.T.; Zhu, F.; Yang, Y.Z.; Yang, F.X.; Hao, J.P.; Chen, S.R.; Hou, Z.C. Genome-wide association study reveals novel loci associated with body size and carcass yields in Pekin ducks. BMC Genom. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- McCaw, Z.R.; Lane, J.M.; Saxena, R.; Redline, S.; Lin, X. Operating characteristics of the rank-based inverse normal transformation for quantitative trait analysis in genome-wide association studies. Biometrics 2020, 76, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Chien, L.C. A rank-based normalization method with the fully adjusted full-stage procedure in genetic association studies. PLoS ONE 2020, 15, e0233847. [Google Scholar] [CrossRef] [PubMed]
- Shalabi, L.A.A.; Shaaban, Z.; Kasasbeh, B. Data Mining: A Preprocessing Engine. J. Comput. Sci. 2006, 2, 735–739. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kendig, K.I.; Baheti, S.; Bockol, M.A.; Drucker, T.M.; Hart, S.N.; Heldenbrand, J.R.; Hernaez, M.; Hudson, M.E.; Kalmbach, M.T.; Klee, E.W.; et al. Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Front. Genet. 2019, 10, 736. [Google Scholar] [CrossRef]
- Davies, R.W.; Flint, J.; Myers, S.; Mott, R. Rapid genotype imputation from sequence without reference panels. Nat. Genet. 2016, 48, 965–969. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Starmer, J.; Martin, E.R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 2008, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.; Cullis, B.R.; Gilmour, A.; Gogel, B.J. ASReml-R Reference Manual; The State of Queensland, Department of Primary Industries and Fisheries: Brisbane, Australia, 2009. [Google Scholar]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Grabe, N. AliBaba2: Context specific identification of transcription factor binding sites. In Silico Biol. 2002, 2, S1–S15. [Google Scholar]
- Wang, G.; Sarkar, A.; Carbonetto, P.; Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Society. Ser. B Stat. Methodol. 2020, 82, 1273–1300. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Dong, S.S.; He, W.M.; Ji, J.J.; Zhang, C.; Guo, Y.; Yang, T.L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 2021, 22, bbaa227. [Google Scholar] [CrossRef]
- Patil, I. Visualizations with statistical details: The ‘ggstatsplot’ approach. J. Open Source Softw. 2021, 6, 3167. [Google Scholar] [CrossRef]
- Isaac, U.; Nosike, R.; Jonas, E.; Helen Chinweoke, E.; Ezenyilimba, B.; Ibe, S. Heritability of Body Weight and Linear Body Measurements of Isa Brown x Local Chicken Genotypes. Int. J. Agric. Environ. Biores. 2023, 8, 90–102. [Google Scholar] [CrossRef]
- Nguyen, T.K.; Lim, J.-H. Tools for Chrysanthemum genetic research and breeding: Is genotyping-by-sequencing (GBS) the best approach? Hortic. Environ. Biotechnol. 2019, 60, 625–635. [Google Scholar] [CrossRef]
- Buffalo, V.; Coop, G. Estimating the genome-wide contribution of selection to temporal allele frequency change. Proc. Natl. Acad. Sci. USA 2020, 117, 20672–20680. [Google Scholar] [CrossRef] [PubMed]
- Wientjes, Y.C.J.; Bijma, P.; van den Heuvel, J.; Zwaan, B.J.; Vitezica, Z.G.; Calus, M.P.L. The long-term effects of genomic selection: 2. Changes in allele frequencies of causal loci and new mutations. Genetics 2023, 225, iyad141. [Google Scholar] [CrossRef] [PubMed]
- Maruoka, M.; Zhang, P.; Mori, H.; Imanishi, E.; Packwood, D.M.; Harada, H.; Kosako, H.; Suzuki, J. Caspase cleavage releases a nuclear protein fragment that stimulates phospholipid scrambling at the plasma membrane. Mol. Cell 2021, 81, 1397–1410.e1399. [Google Scholar] [CrossRef] [PubMed]
- Terakado, A.P.N.; Costa, R.B.; de Camargo, G.M.F.; Irano, N.; Bresolin, T.; Takada, L.; Carvalho, C.V.D.; Oliveira, H.N.; Carvalheiro, R.; Baldi, F.; et al. Genome-wide association study for growth traits in Nelore cattle. Anim. Int. J. Anim. Biosci. 2018, 12, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Xia, J.; Chang, T.; Wang, X.; Xu, L.; Zhang, L.; Gao, X.; Chen, Y.; Li, J.; Gao, H. Genome-wide association study reveals candidate genes associated with body measurement traits in Chinese Wagyu beef cattle. Anim. Genet. 2019, 50, 386–390. [Google Scholar] [CrossRef]
- Lindholm-Perry, A.K.; Kuehn, L.A.; Smith, T.P.; Ferrell, C.L.; Jenkins, T.G.; Freetly, H.C.; Snelling, W.M. A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle(1). Anim. Genet. 2012, 43, 216–219. [Google Scholar] [CrossRef]
- Porto Neto, L.R.; Bunch, R.J.; Harrison, B.E.; Barendse, W. Variation in the XKR4 gene was significantly associated with subcutaneous rump fat thickness in indicine and composite cattle. Anim. Genet. 2012, 43, 785–789. [Google Scholar] [CrossRef]
- Yong, R.; Xiang, C.; Gui-gang, T.; Dong-wei, A.N.; Qi-shun, Z.O.U. Correlation analysis of XKR4 gene polymorphism and growth traits in F3 generation Boza goats. J. South. Agric. 2022, 53, 1752–1758. [Google Scholar] [CrossRef]
- Zou, L.; Menon, R.K. A member of the CTF/NF-1 transcription factor family regulates murine growth hormone receptor gene promoter activity. Endocrinology 1995, 136, 5236–5239. [Google Scholar] [CrossRef] [PubMed]
- Reumann, M.K.; Strachna, O.; Lukashova, L.; Verdelis, K.; Donnelly, E.; Boskey, A.L.; Mayer-Kuckuk, P. Early growth response gene 1 regulates bone properties in mice. Calcif. Tissue Int. 2011, 89, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Toan, N.K.; Tai, N.C.; Kim, S.A.; Ahn, S.G. Soluble Klotho regulates bone differentiation by upregulating expression of the transcription factor EGR-1. FEBS Lett. 2020, 594, 290–300. [Google Scholar] [CrossRef]
- Wang, F.; Meyer, N.J.; Walley, K.R.; Russell, J.A.; Feng, R. Causal Genetic Inference Using Haplotypes as Instrumental Variables. Genet. Epidemiol. 2016, 40, 35–44. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Traits | (h2) 5 | Means | Min | Max | CV 6 | N 7 |
|---|---|---|---|---|---|---|
| BL (cm) 1 | 0.45 ± 0.09 | 26.49 ± 1.16 | 23.30 | 29.70 | 4.38 | 641 |
| KBL (cm) 2 | 0.31 ± 0.08 | 13.58 ± 0.47 | 12.10 | 15.00 | 3.48 | 641 |
| NL (cm) 3 | 0.37 ± 0.08 | 21.06 ± 0.94 | 18.70 | 23.90 | 4.47 | 641 |
| BrW (cm) 4 | 0.40 ± 0.09 | 12.00 ± 0.41 | 10.80 | 13.20 | 3.46 | 642 |
| Traits | Lead SNP | p-Value | Ref 1 | Alt 2 | MAF 3 | Gene | Consequence 4 |
|---|---|---|---|---|---|---|---|
| BL | 2:41045610 | 2.02 × 10−12 | C | T | 0.457 | XKR4 | Intron variant |
| KBL | 2:41041078 | 1.21 × 10−7 | A | G | 0.417 | XKR4 | Intron variant |
| NL | 23:6102898 | 5.24 × 10−7 | G | A | 0.441 | - | Intergenic variant |
| BrW | 2:40713142 | 4.00 × 10−7 | G | A | 0.452 | Pseudogene | Downstream variant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.-Z.; Zhou, J.; Yang, F.-X.; Hao, J.-P.; Hou, Z.-C.; Zhu, F. Genome-Wide Association Analysis Identifies Important Haplotypes and Candidate Gene XKR4 for Body Size Traits in Pekin Ducks. Animals 2024, 14, 2349. https://doi.org/10.3390/ani14162349
Yu J-Z, Zhou J, Yang F-X, Hao J-P, Hou Z-C, Zhu F. Genome-Wide Association Analysis Identifies Important Haplotypes and Candidate Gene XKR4 for Body Size Traits in Pekin Ducks. Animals. 2024; 14(16):2349. https://doi.org/10.3390/ani14162349
Chicago/Turabian StyleYu, Jiang-Zhou, Jun Zhou, Fang-Xi Yang, Jin-Ping Hao, Zhuo-Cheng Hou, and Feng Zhu. 2024. "Genome-Wide Association Analysis Identifies Important Haplotypes and Candidate Gene XKR4 for Body Size Traits in Pekin Ducks" Animals 14, no. 16: 2349. https://doi.org/10.3390/ani14162349