
Comparative Analysis of Gut Bacterial Diversity in Wild and Domestic Yaks on the Qinghai–Tibetan Plateau


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The Experimental Site, Animals, and Management
2.2. Determination of Nutritional Composition of Experimental Forages
2.3. DNA Extraction, PCR Amplification, and MiSeq Sequencing of 16S rRNA Gene Amplicons
2.4. Sequencing and Data Processing
2.5. Statistical Analysis
3. Results
3.1. Analysis of Sequencing Data and Bacterial Diversity
3.2. Alpha Diversity
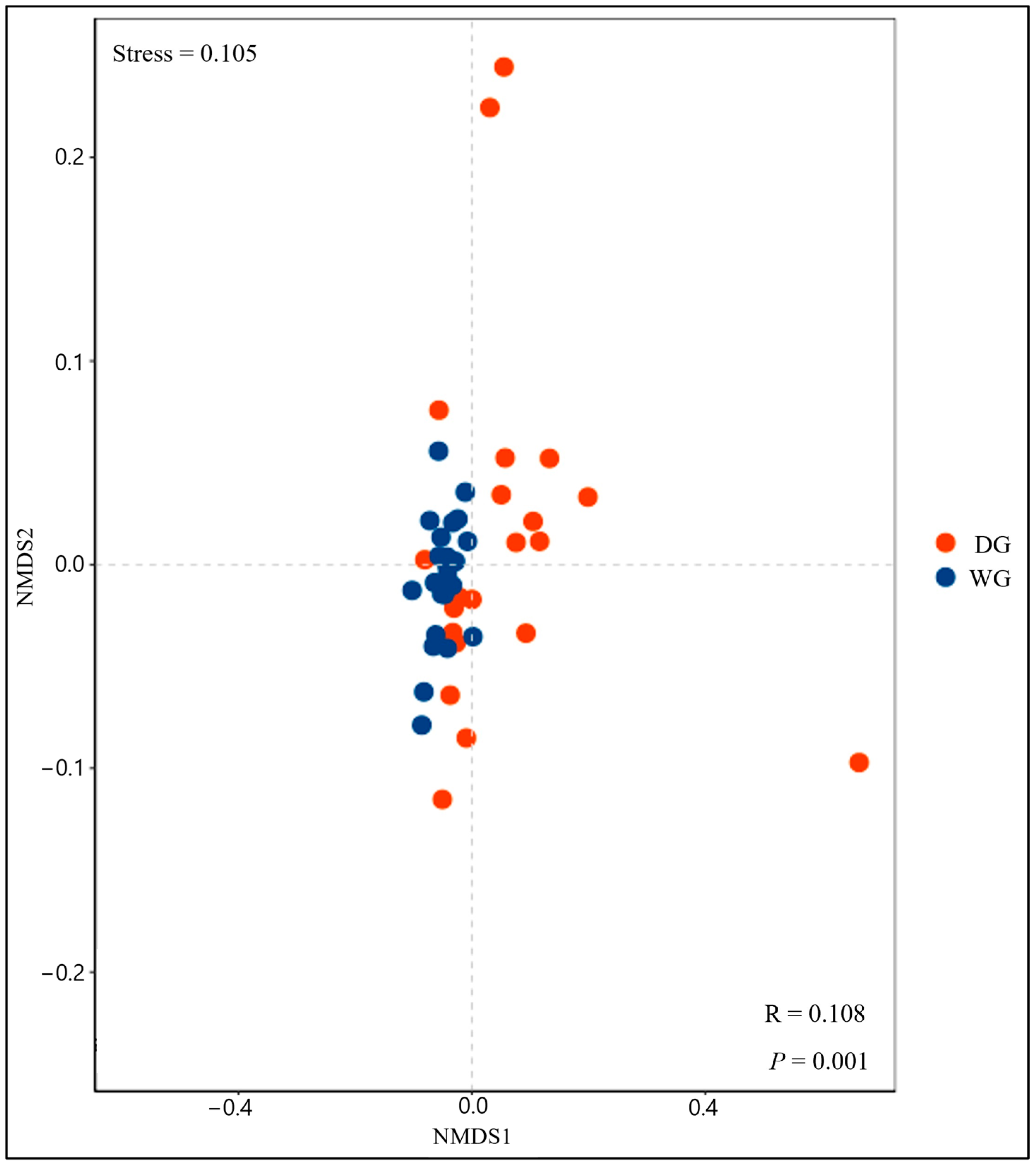
3.3. Beta Diversity
3.4. Gut Bacterial Composition
3.5. Relative Abundance of Bacterial Families
3.6. Relative Abundance of Bacterial Genera
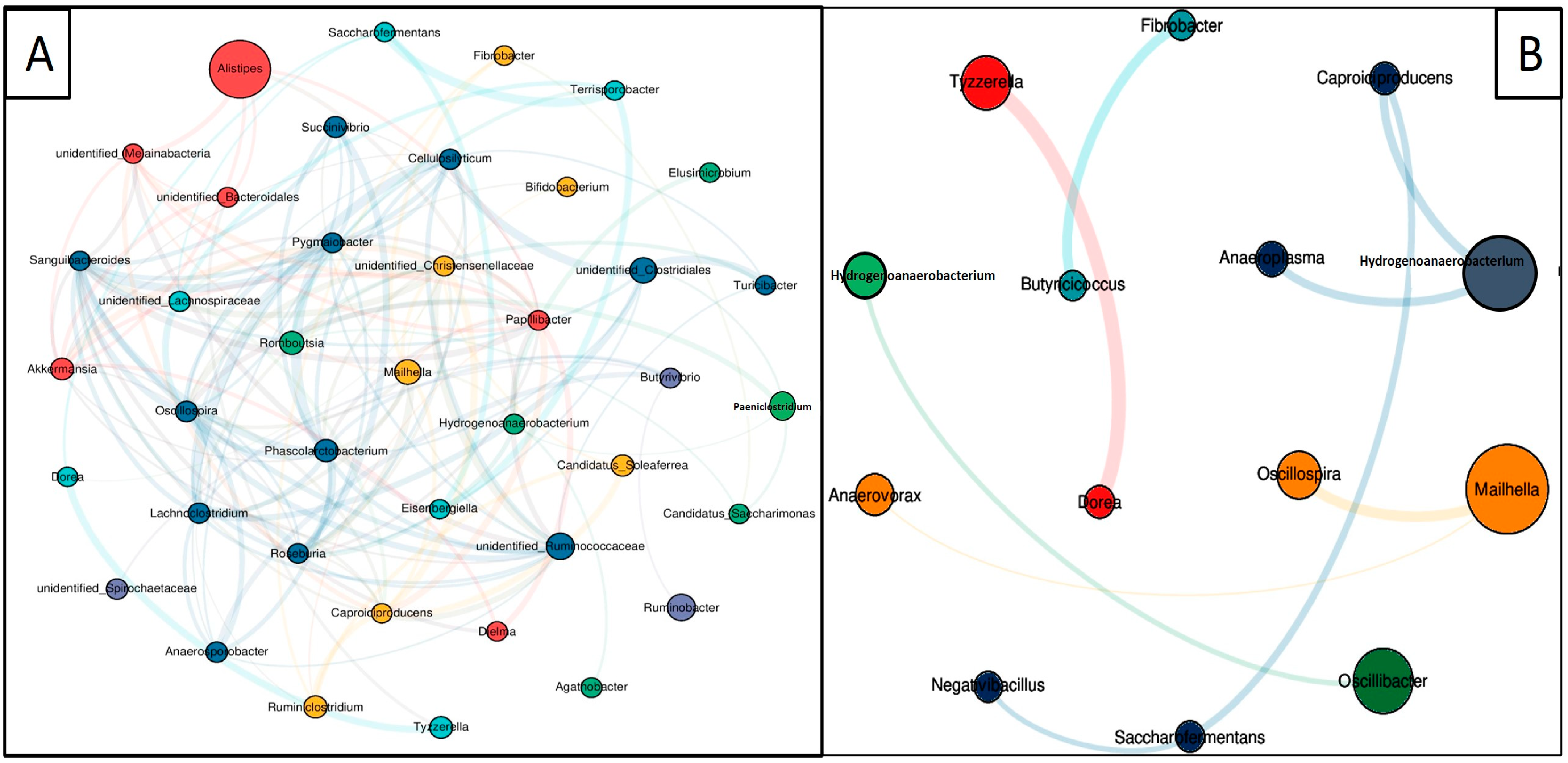
3.7. Co-Occurrence Network Analysis
4. Discussion
4.1. Forage Quality and Microbial Diversity
4.2. Differences in Gut Mircobiota
4.3. Phylum-Level Bacterial Composition
4.4. Family-Level Bacterial Composition
4.5. Genus-Level Bacterial Composition
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Zhang, Z.; Li, B.; Hao, W.; Yin, W.; Ai, S.; Han, J.; Rujing Wang, R.; Duan, Z. Depicting Fecal Microbiota Characteristic in Yak, Cattle, Yak-Cattle Hybrid and Tibetan Sheep in Different Eco-Regions of Qinghai-Tibetan Plateau. Microbiol. Spectr. 2022, 10, e00021-22. [Google Scholar] [CrossRef]
- Wang, H.; Long, R.; Liang, J.B.; Guo, X.; Ding, L.; Shang, Z. Comparison of nitrogen metabolism in yak (Bos grunniens) and indigenous cattle (Bos taurus) on the Qinghai-Tibetan Plateau. Asian-Australasian J. Anim. Sci. 2011, 24, 766–773. [Google Scholar] [CrossRef]
- Ren, M.; Song, J.K.; Yang, F.; Zou, M.; Wang, P.X.; Wang, D.; Zhang, H.J.; Zhao, G.H.; Lin, Q. First genotyping of Blastocystis in yaks from Qinghai Province, northwestern China. Parasites Vectors 2019, 12, 171. [Google Scholar] [CrossRef]
- Ma, J.; Shah, A.M.; Wang, Z.; Hu, R.; Zou, H.; Wang, X.; Cao, G.; Peng, Q.; Xue, B.; Wang, L.; et al. Comparing the gastrointestinal barrier function between growth-retarded and normal yaks on the Qinghai-Tibetan Plateau. PeerJ 2020, 8, e9851. [Google Scholar] [CrossRef]
- Hu, R.; Zou, H.; Wang, Z.; Cao, B.; Peng, Q.; Jing, X.; Wang, Y.; Shao, Y.; Pei, Z.; Zhang, X.; et al. Nutritional interventions improved rumen functions and promoted compensatory growth of growth-retarded yaks as revealed by integrated transcripts and microbiome analyses. Front. Microbiol. 2019, 10, 318. [Google Scholar] [CrossRef]
- Xu, T.; Xu, S.; Hu, L.; Zhao, N.; Liu, Z.; Ma, L.; Liu, H.; Zhao, X. Effect of dietary types on feed intakes, growth performance and economic benefit in tibetan sheep and yaks on the qinghai-tibet plateau during cold season. PLoS ONE 2017, 12, e0169187. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Teotia, U.; Aswal, A. Antihypertensive Property of Yak Milk Caseinates Hydrolyzed with Different Proteases. Int. J. Livest. Res. 2013, 3, 130. [Google Scholar] [CrossRef]
- Nikkhah, A. Equidae, Camel, and Yak Milks as Functional Foods: A Review. J. Nutr. Food Sci. 2011, 1, 116. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, D.; Wang, L.; Hao, J.; Wang, J.; Zhou, X.; Wang, W.; Qiu, Q.; Huang, X.; Zhou, J.; et al. Convergent Evolution of Rumen Microbiomes in High-Altitude Mammals. Curr. Biol. 2016, 26, 1873–1879. [Google Scholar] [CrossRef]
- Liu, H.; Li, Z.; Pei, C.; Degen, A.; Hao, L.; Cao, X.; Liu, H.; Zhou, J.; Long, R. A comparison between yaks and Qaidam cattle in in vitro rumen fermentation, methane emission, and bacterial community composition with poor quality substrate. Anim. Feed Sci. Technol. 2022, 291, 115395. [Google Scholar] [CrossRef]
- Zi, X.D. Reproduction in female yaks (Bos grunniens) and opportunities for improvement. Theriogenology 2003, 59, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhu, Y.; Wang, Z.; Yu, X.; Hu, R.; Wang, X.; Cao, G.; Zou, H.; Shah, A.M.; Peng, Q.; et al. Comparing the Bacterial Community in the Gastrointestinal Tracts between Growth-Retarded and Normal Yaks on the Qinghai–Tibetan Plateau. Front. Microbiol. 2020, 11, 600516. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Vinitchaikul, P.; Deng, M.; Zhang, G.; Sun, L.; Wang, H.; Gou, X.; Mao, H.; Yang, S. Exploration of the effects of altitude change on bacteria and fungi in the rumen of yak (Bos grunniens). Arch. Microbiol. 2021, 203, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Celi, P.; Verlhac, V.; Pérez Calvo, E.; Schmeisser, J.; Kluenter, A.M. Biomarkers of gastrointestinal functionality in animal nutrition and health. Anim. Feed Sci. Technol. 2019, 250, 9–31. [Google Scholar] [CrossRef]
- Parker, A.; Lawson, M.A.E.; Vaux, L.; Pin, C. Host-microbe interaction in the gastrointestinal tract. Environ. Microbiol. 2018, 20, 2337–2353. [Google Scholar] [CrossRef] [PubMed]
- Yeoman, C.J.; White, B.A. Gastrointestinal tract microbiota and probiotics in production animals. Annu. Rev. Anim. Biosci. 2014, 2, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Matthews, C.; Crispie, F.; Lewis, E.; Reid, M.; O’Toole, P.W.; Cotter, P.D. The rumen microbiome: A crucial consideration when optimising milk and meat production and nitrogen utilisation efficiency. Gut Microbes 2019, 10, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carrio, J.; Salazar, N.; Margolles, A.; González, S.; Gueimonde, M.; de los Reyes-Gavilán, C.G.; Suárez, A. Free fatty acids profiles are related to gut microbiota signatures and short-chain fatty acids. Front. Immunol. 2017, 8, 823. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.L.R.; Phillips, M.W. The role of anaerobic gut fungi in ruminants. Nutr. Res. Rev. 1998, 11, 133–168. [Google Scholar] [CrossRef]
- Yohe, T.T.; Schramm, H.; White, R.R.; Hanigan, M.D.; Parsons, C.L.M.; Tucker, H.L.M.; Enger, B.D.; Hardy, N.R.; Daniels, K.M. Form of calf diet and the rumen. II: Impact on volatile fatty acid absorption. J. Dairy Sci. 2019, 102, 8502–8512. [Google Scholar] [CrossRef]
- Black, J.L.; Davison, T.M.; Box, I. Methane Emissions from Ruminants in Australia: Mitigation Potential and Applicability of Mitigation Strategies. Animals 2021, 11, 951. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Lee, S. Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Yang, C.; Zhang, J.; Kalwar, Q.; Liang, Z.; Li, C.; Du, M.; Yan, P.; Long, R.; Han, J.; et al. Effects of Dietary Energy Levels on Rumen Fermentation, Microbial Diversity, and Feed Efficiency of Yaks (Bos grunniens). Front. Microbiol. 2020, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H.; Abecia, L.; Angarita, E.; Aravena, P.; Arenas, G.N.; et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Chen, Y.; Liu, J.; Zhang, C.; Irving, B.; Fitzsimmons, C.; Plastow, G.; Guan, L.L. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, A.M.; Liebetrau, J.; Pröter, J.; Kleinsteuber, S. Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl. Microbiol. Biotechnol. 2013, 97, 5161–5174. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhou, Z.; Ren, L.; Shi, F.; Can, M.; Chai, S.; Meng, Q. Ruminal bacterial diversity of yaks (Bos grunniens) fed by grazing or indoor regime on the Tibetan Plateau by analysis of 16S rRNA gene libraries. Ital. J. Anim. Sci. 2015, 14, 621–627. [Google Scholar] [CrossRef]
- Ma, L.; Xu, S.; Liu, H.; Xu, T.; Hu, L.; Zhao, N.; Han, X.; Zhang, X. Yak rumen microbial diversity at different forage growth stages of an alpine meadow on the Qinghai-Tibet Plateau. PeerJ 2019, 7, e7645. [Google Scholar] [CrossRef]
- Tajima, K.; Aminov, R.I.; Nagamine, T.; Ogata, K.; Nakamura, M.; Matsui, H.; Benno, Y. Rumen bacterial diversity as determined by sequence analysis of 16S rDNA libraries. FEMS Microbiol. Ecol. 1999, 29, 159–169. [Google Scholar] [CrossRef]
- Huang, C.; Ge, F.; Yao, X.; Guo, X.; Bao, P.; Ma, X.; Wu, X.; Chu, M.; Yan, P.; Liang, C. Microbiome and Metabolomics Reveal the Effects of Different Feeding Systems on the Growth and Ruminal Development of Yaks. Front. Microbiol. 2021, 12, 682989. [Google Scholar] [CrossRef]
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for Dietary Fiber, Neutral Detergent Fiber, and Nonstarch Polysaccharides in Relation to Animal Nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef] [PubMed]
- Thiex, N.; Novotny, L.; Crawford, A. Determination of ash in animal feed: AOAC Official Method 942.05 revisited. J. AOAC Int. 2012, 95, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, H.; Wright, C.L.; Li, X.; Lin, Q.; Hwang, C.; Wang, S.; Thimmapuram, J.; Kamagata, Y.; Liu, W.T. Analysis of 16S rRNA amplicon sequencing options on the roche/454 next-generation titanium sequencing platform. PLoS ONE 2011, 6, e0025263. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. correspondence QIIME allows analysis of high- throughput community sequencing data Intensity normalization improves color calling in SOLiD sequencing. Nat. Publ. Gr. 2010, 7, 335–336. [Google Scholar]
- Li, H.; Qu, J.; Li, T.; Li, J.; Lin, Q.; Li, X. Pika population density is associated with the composition and diversity of gut microbiota. Front. Microbiol. 2016, 7, 758. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “all-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Loor, J.J.; Elolimy, A.A.; McCann, J.C. Dietary impacts on rumen microbiota in beef and dairy production. Anim. Front. 2016, 6, 22–29. [Google Scholar] [CrossRef]
- Promkot, C.; Wanapat, M. Ruminal degradation and intestinal digestion of crude protein of tropical protein resources using nylon bag technique and three-step in vitro procedure in dairy cattle. Livest. Res. Rural Dev. 2003, 15, 1–7. [Google Scholar]
- Mupangwa, J.F.; Ngongoni, N.T.; Topps, J.H.; Ndlovu, P. Chemical composition and dry matter degradability profiles of forage legumes Cassia rotundifolia cv. Wynn, Lablab purpureus cv. Highworth and Macroptilium atropurpureum cv. Siratro at 8 weeks of growth (pre-anthesis). Anim. Feed Sci. Technol. 1997, 69, 167–178. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Preston, T.R. Rumen environment and feed degradability in swamp buffaloes fed different supplements. Livest. Res. Rural Dev. 1999, 11, 10–16. [Google Scholar]
- Latham, E.A.; Weldon, K.K.; Wickersham, T.A.; Coverdale, J.A.; Pinchak, W.E. Responses in the rumen microbiome of Bos taurus and indicus steers fed a low-quality rice straw diet and supplemented protein. J. Anim. Sci. 2018, 96, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Belanche, A.; Doreau, M.; Edwards, J.E.; Moorby, J.M.; Pinloche, E.; Newbold, C.J. Shifts in the Rumen Microbiota Due to the Type of Carbohydrate and Level of Protein Ingested by Dairy Cattle Are Associated with Changes in Rumen Fermentation. J. Nutr. 2012, 142, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Kittelmann, S.; Rogers, C.W.; Gee, E.K.; Bolwell, C.F.; Bermingham, E.N.; Thomas, D.G. Faecal microbiota of forage-fed horses in new zealand and the population dynamics of microbial communities following dietary change. PLoS ONE 2014, 9, e0112846. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Wang, J.K.; Wu, Y.M.; Liu, J.X. Effects of chemical treatments of rice straw on rumen fermentation characteristics, fibrolytic enzyme activities and populations of liquid- and solid-associated ruminal microbes in vitro. Anim. Feed Sci. Technol. 2008, 141, 1–14. [Google Scholar] [CrossRef]
- Guo, N.; Wu, Q.; Shi, F.; Niu, J.; Zhang, T.; Degen, A.A.; Fang, Q.; Ding, L.; Shang, Z.; Zhang, Z.; et al. Seasonal dynamics of diet–gut microbiota interaction in adaptation of yaks to life at high altitude. NPJ Biofilms Microbiomes 2021, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Zhou, Z.; Guan, J.; Xia, B.; Luo, X.; Yang, Y.; Fu, Y.; Sun, Q. Dynamic changes of yak (Bos grunniens) gut microbiota during growth revealed by polymerase chain reaction-denaturing gradient gel electrophoresis and metagenomics. Asian-Australas. J. Anim. Sci. 2017, 30, 957–966. [Google Scholar] [CrossRef]
- Liu, C.; Wu, H.; Liu, S.; Chai, S.; Meng, Q.; Zhou, Z. Dynamic alterations in yak rumen bacteria community and metabolome characteristics in response to feed type. Front. Microbiol. 2019, 10, 1116. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Xue, M.; Liu, J. Composition of Rumen Bacterial Community in Dairy Cows with Different Levels of Somatic Cell Counts. Front. Microbiol. 2018, 9, 3217. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; Chai, Z.; Zhang, C.; Zhang, Q.; Zhu, Y.; Cao, H.; Zhong, J.; Ji, Q. Comparing the microbial community in four stomach of dairy cattle, yellow cattle and three yak herds in qinghai-tibetan plateau. Front. Microbiol. 2019, 10, 1547. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Ali Ahmad, A.; Bo Zhang, J.; Liang, Z.; Yang, C.; Kalwar, Q.; Shah, T.; Du, M.; Muhammad, I.; Zheng, J.; Yan, P.; et al. Dynamics of rumen bacterial composition of yak (Bos grunniens) in response to dietary supplements during the cold season. PeerJ 2021, 9, e11520. [Google Scholar] [CrossRef]
- Xue, D.; Chen, H.; Chen, F.; He, Y.; Zhao, C.; Zhu, D.; Zeng, L.; Li, W. Analysis of the rumen bacteria and methanogenic archaea of yak (Bos grunniens) steers grazing on the Qinghai-Tibetan Plateau. Livest. Sci. 2016, 188, 61–71. [Google Scholar] [CrossRef]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e0085423. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Patel, A.K.; Parmar, N.R.; Patel, A.B.; Reddy, B.; Joshi, C.G. Characterization of the rumen microbiome of Indian Kankrej cattle (Bos indicus) adapted to different forage diet. Appl. Microbiol. Biotechnol. 2014, 98, 9749–9761. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, Y.; Luo, Y. Bacterial community in the rumen of tibetan sheep and gansu alpine fine-wool sheep grazing on the Qinghai-Tibetan Plateau, China. J. Gen. Appl. Microbiol. 2017, 63, 122–130. [Google Scholar] [CrossRef]
- Han, X.; Yang, Y.; Yan, H.; Wang, X.; Qu, L.; Chen, Y. Rumen bacterial diversity of 80 to 110-day-Old goats using 16s rRNA sequencing. PLoS ONE 2015, 10, e0117811. [Google Scholar] [CrossRef]
- Pitta, D.W.; Pinchak, W.E.; Dowd, S.E.; Osterstock, J.; Gontcharova, V.; Youn, E.; Dorton, K.; Yoon, I.; Min, B.R.; Fulford, J.D.; et al. Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb. Ecol. 2010, 59, 511–522. [Google Scholar] [CrossRef]
- Guo, X.; Xia, X.; Tang, R.; Wang, K. Real-time PCR quantification of the predominant bacterial divisions in the distal gut of Meishan and Landrace pigs. Anaerobe 2008, 14, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Chen, H.; Zhao, X.; Xu, S.; Hu, L.; Xu, T.; Jiang, L.; Zhan, W. Rumen prokaryotic communities of ruminants under different feeding paradigms on the Qinghai-Tibetan Plateau. Syst. Appl. Microbiol. 2017, 40, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, K.; Wang, Z.; Bai, X.; Peng, Q.; Jin, L. Bacterial community diversity associated with different utilization efficiencies of nitrogen in the gastrointestinal tract of goats. Front. Microbiol. 2019, 10, 239. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012, 6, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Ziemer, C.J. Newly cultured bacteria with broad diversity isolated from eight-week continuous culture enrichments of cow feces on complex polysaccharides. Appl. Environ. Microbiol. 2014, 80, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Purushe, J.; Fouts, D.E.; Morrison, M.; White, B.A.; Mackie, R.I.; Coutinho, P.M.; Henrissat, B.; Nelson, K.E. Comparative Genome Analysis of Prevotella ruminicola and Prevotella bryantii: Insights into Their Environmental Niche. Microb. Ecol. 2010, 60, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, M.; Xue, C.; Zhu, W.; Mao, S. Characterization and comparison of the temporal dynamics of ruminal bacterial microbiota colonizing rice straw and alfalfa hay within ruminants. J. Dairy Sci. 2016, 99, 9668–9681. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Liu, Z.; Yu, Z.; Zhu, W. Monensin and nisin affect rumen fermentation and microbiota differently in vitro. Front. Microbiol. 2017, 8, 1111. [Google Scholar] [CrossRef]
- Hamlin, L.J.; Hungate, R.E. Culture and physiology of a starch-digesting bacterium (Bacteroides amylophilus n. sp.) from the bovine rumen. J. Bacteriol. 1956, 72, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Y.; Zhang, Y.; Wang, L. The Effects of Different Concentrate-to-Forage Ratio Diets on Rumen Bacterial Microbiota and the Structures of Holstein Cows during the Feeding Cycle. Animals 2020, 10, 957. [Google Scholar] [CrossRef] [PubMed]
- Ramšak, A.; Peterka, M.; Tajima, K.; Martin, J.C.; Wood, J.; Johnston, M.E.A.; Aminov, R.I.; Flint, H.J.; Avguštin, G. Unravelling the genetic diversity of ruminal bacteria belonging to the CFB phylum. FEMS Microbiol. Ecol. 2000, 33, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.M.; Dehority, B.A. Synergism in Degradation and Utilization of Intact Forage Cellulose, Hemicellulose, and Pectin by Three Pure Cultures of Ruminal Bacteria. Appl. Environ. Microbiol. 1989, 55, 2247–2250. [Google Scholar] [CrossRef]
- Gulino, L.M.; Ouwerkerk, D.; Kang, A.Y.H.; Maguire, A.J.; Kienzle, M.; Klieve, A.V. Shedding Light on the Microbial Community of the Macropod Foregut Using 454-Amplicon Pyrosequencing. PLoS ONE 2013, 8, e61463. [Google Scholar] [CrossRef]
- Wallace, R.J.; McKain, N.; Broderick, G.A.; Rode, L.M.; Walker, N.D.; Newbold, C.J.; Kopecny, J. Peptidases of the rumen bacterium, Prevotella ruminicola. Anaerobe 1997, 3, 35–42. [Google Scholar] [CrossRef]
- Shah, H.N.; Collins, M.D. Proposal to restrict the genus Bacteroides (Castellani and Chalmers) to Bacteroides fragilis and closely related species. Int. J. Syst. Bacteriol. 1989, 39, 85–87. [Google Scholar] [CrossRef]
- Windey, K.; de Preter, V.; Verbeke, K. Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 2012, 56, 184–196. [Google Scholar] [CrossRef]
- Wrzosek, L.; Miquel, S.; Noordine, M.L.; Bouet, S.; Chevalier-Curt, M.J.; Robert, V.; Philippe, C.; Bridonneau, C.; Cherbuy, C.; Robbe-Masselot, C.; et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Mach, N.; Lepage, P.; Levenez, F.; Denis, C.; Lemonnier, G.; Leplat, J.J.; Billon, Y.; Berri, M.; Doré, J.; et al. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J. 2016, 10, 2973–2977. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Forage in DG | Forage in WG | SEM | p-Value |
|---|---|---|---|---|
| Dry matter | 97.00% | 96.96% | 0.047 | 0.652 |
| Organic matter | 92.58% | 92.63% | 0.035 | 0.716 |
| Ash | 7.30% | 7.29% | 0.009 | 0.345 |
| Crude protein | 15.10% | 7.30% | 5.025 | 0.001 |
| Neutral detergent fiber | 53.78% | 57.68% | 2.757 | 0.001 |
| Acid detergent fiber | 27.8% | 30.81% | 2.133 | 0.012 |
| Ether extract | 1.37% | 1.5% | 0.091 | 0.029 |
| Total carbohydrates | 76.53% | 77.43% | 0.636 | 0.095 |
| Non-fiber carbohydrates | 22.76% | 22.1% | 0.471 | 0.034 |
| Hemicellulose | 26.13% | 26.33% | 0.141 | 0.467 |
| Taxonomy | DG | WG | SEM | p-Value |
|---|---|---|---|---|
| Bacteroidetes | 0.91 b | 1.08 a | 0.119 | 0.052 |
| Firmicutes | 1.12 a | 0.89 b | 0.162 | 0.024 |
| Proteobacteria | 0.93 | 1.06 | 0.088 | 0.505 |
| Elusimicrobia | 1.07 a | 0.93 b | 0.098 | 0.032 |
| Fusobacteria | 1.18 a | 0.84 b | 0.246 | 0.001 |
| Kiritimatiellaeota | 1.08 a | 0.92 b | 0.114 | 0.042 |
| Lentisphaerae | 1.15 | 0.86 | 0.198 | 0.0002 |
| Spirochaetes | 0.81 | 1.22 | 0.293 | 0.108 |
| Fibrobacteres | 0.86 b | 1.15 a | 0.202 | 0.059 |
| Tenericutes | 0.93 | 1.07 | 0.098 | 0.297 |
| Verrucomicrobia | 1.09 | 0.91 | 0.127 | 0.096 |
| Melainabacteria | 1.06 | 0.94 | 0.083 | 0.127 |
| Taxonomy | DG | WG | SEM | p-Value |
|---|---|---|---|---|
| Ruminococcaceae | 1.22 a | 0.81 b | 0.286 | 0.0001 |
| Succinivibrionaceae | 0.89 | 1.11 | 0.160 | 0.562 |
| Prevotellaceae | 0.85 b | 1.16 a | 0.219 | 0.0003 |
| Lachnospiraceae | 0.82 | 1.21 | 0.271 | 7.889 |
| Rikenellaceae | 0.97 | 1.03 | 0.042 | 0.372 |
| Bacteroidaceae | 0.96 | 1.04 | 0.056 | 0.161 |
| Muribaculaceae | 0.84 b | 1.18 a | 0.244 | 0.043 |
| Taxonomy | DG | WG | SEM | p-Value |
|---|---|---|---|---|
| Succinivibrio | 0.78 | 1.26 | 0.340 | 0.060 |
| Ruminobacter | 0.92 | 1.07 | 0.103 | 0.742 |
| Alloprevotella | 0.89 | 1.11 | 0.160 | 0.068 |
| Faecalibacterium | 0.90 | 1.10 | 0.144 | 0.252 |
| Bacteroides | 0.96 | 1.04 | 0.056 | 0.161 |
| Anaerovibrio | 0.89 | 1.12 | 0.162 | 0.448 |
| Alistipes | 1.02 | 0.97 | 0.030 | 0.487 |
| Mailhella | 1.07 a | 0.93 b | 0.099 | 0.018 |
| Tyzzerella | 1.03 | 0.96 | 0.047 | 0.105 |
| S. No | Network Attributes | DG | WG |
|---|---|---|---|
| 1 | Nodes | 40 | 14 |
| 2 | Edges | 146 | 9 |
| 3 | Average degree | 7.3 | 1.285714286 |
| 4 | Average path length | 2.457692308 | 1.6875 |
| 5 | Network diameter | 4 | 3 |
| 6 | Clustering coefficient | 0.577793009 | 0 |
| 7 | Density | 0.187179487 | 0.098901099 |
| 8 | Heterogeneity | 0.72033543 | 0.364627846 |
| 9 | Centralization | 0.223076923 | 0.054945055 |
| 10 | Positive correlation | 58.90% | 66.66% |
| 11 | Negative correlation | 41.10% | 33.34% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, T.; Guo, X.; Song, Y.; Fang, Y.; Ding, L. Comparative Analysis of Gut Bacterial Diversity in Wild and Domestic Yaks on the Qinghai–Tibetan Plateau. Animals 2024, 14, 2380. https://doi.org/10.3390/ani14162380
Shah T, Guo X, Song Y, Fang Y, Ding L. Comparative Analysis of Gut Bacterial Diversity in Wild and Domestic Yaks on the Qinghai–Tibetan Plateau. Animals. 2024; 14(16):2380. https://doi.org/10.3390/ani14162380
Chicago/Turabian StyleShah, Tariq, Xusheng Guo, Yongwu Song, Yonggui Fang, and Luming Ding. 2024. "Comparative Analysis of Gut Bacterial Diversity in Wild and Domestic Yaks on the Qinghai–Tibetan Plateau" Animals 14, no. 16: 2380. https://doi.org/10.3390/ani14162380
APA StyleShah, T., Guo, X., Song, Y., Fang, Y., & Ding, L. (2024). Comparative Analysis of Gut Bacterial Diversity in Wild and Domestic Yaks on the Qinghai–Tibetan Plateau. Animals, 14(16), 2380. https://doi.org/10.3390/ani14162380