

Exploring the Genetic Landscape of Vitiligo in the Pura Raza Español Horse: A Genomic Perspective

 , ,
, ,  , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Phenotypes
2.2. Genotyping and Quality Control
2.3. Weighted Single-Step Genome-Wide Association Analysis
2.4. Functional Analysis of Significant Genomic Regions
3. Results
3.1. Descriptive Statistics
3.2. Genome-Wide Association Studies
4. Discussion
4.1. Prevalence and Genetics Parameters
4.2. Genomic Association with Vitiligo
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taieb, A.; Alomar, A.; Böhm, M.; Dell’Anna, M.L.; De Pase, A.; Eleftheriadou, V.; Ezzedine, K.; Gauthier, Y.; Gawkrodger, D.J.; Jouary, T.; et al. Guidelines for the management of vitiligo: The European Dermatology Forum consensus. Br. J. Dermatol. 2013, 168, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, G.F.; Gomaa, A.H.; Al-Dhubaibi, M.S. Highlights in pathogenesis of vitiligo. World J. Clin. Cases 2015, 3, 221–230. [Google Scholar] [CrossRef]
- Bibeau, K.; Pandya, A.G.; Ezzedine, K.; Jones, H.; Gao, J.; Lindley, A.; Harris, J.E. Vitiligo prevalence and quality of life among adults in Europe, Japan and the USA. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1831–1844. [Google Scholar] [CrossRef] [PubMed]
- Tham, H.L.; Linder, K.E.; Olivry, T. Autoimmune diseases affecting skin melanocytes in dogs, cats and horses: Vitiligo and the uveodermatological syndrome: A comprehensive review. BMC Vet. Res. 2019, 15, 251. [Google Scholar] [CrossRef]
- Lindgren, G.; Naboulsi, R.; Frey, R.; Solé, M. Genetics of Skin Disease in Horses. Vet. Clin. N. Am. Equine Pract. 2020, 36, 323–339. [Google Scholar] [CrossRef]
- Sánchez-Guerrero, M.J.; Solé, M.; Azor, P.J.; Sölkner, J.; Valera, M. Genetic and environmental risk factors for vitiligo and melanoma in Pura Raza Español horses. Equine Vet. J. 2019, 51, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Druml, T.; Brem, G.; Velie, B.; Lindgren, G.; Horna, M.; Ricard, A.; Grilz-Seger, G. Equine vitiligo-like depigmentation in grey horses is related to genes involved in immune response and tumor metastasis. BMC Vet. Res. 2021, 17, 336. [Google Scholar] [CrossRef]
- Curik, I.; Druml, T.; Seltenhammer, M.; Sundström, E.; Pielberg, G.R.; Andersson, L.; Sölkner, J. Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses. PLoS Genet. 2013, 9, e1003248. [Google Scholar] [CrossRef]
- Shen, C.; Gao, J.; Sheng, Y.; Dou, J.; Zhou, F.; Zheng, X.; Ko, R.; Tang, X.; Zhu, C.; Yin, X.; et al. Genetic Susceptibility to Vitiligo: GWAS Approaches for Identifying Vitiligo Susceptibility Genes and Loci. Front. Genet. 2016, 7, 3. [Google Scholar] [CrossRef]
- Gupta, I.; Narang, A.; Singh, P.; Manchanda, V.; Khanna, S.; Mukerji, M.; Natarajan, V.T.; Dash, D. VitiVar: A locus specific database of vitiligo associated genes and variations. Gene 2019, 721, 100018. [Google Scholar] [CrossRef]
- Jin, Y.; Andersen, G.; Yorgov, D.; Ferrara, T.M.; Ben, S.; Brownson, K.M.; Holland, P.J.; Birlea, S.A.; Siebert, J.; Hartmann, A.; et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet. 2016, 48, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Yu, S.; Ding, Y.; Liang, J.; Halifu, Y.; Xiang, F.; Zhang, D.; Wang, H.; Hu, W.; Li, T.; et al. Identification of key genes and pathways involved in vitiligo development based on integrated analysis. Medicine 2020, 99, e21297. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.; Mitra, S.; Saha, A.; Ganguly, K.; Pyne, T.; Sengupta, M. A comprehensive meta-analysis and prioritization study to identify vitiligo associated coding and non-coding SNV candidates using web-based bioinformatics tools. Sci. Rep. 2022, 12, 14543. [Google Scholar] [CrossRef]
- Cheng, L.; Liang, B.; Tang, X.-F.; Cai, X.-Y.; Cheng, H.; Zheng, X.-D.; Zheng, J.; Wang, M.-W.; Zhu, J.; Zhou, F.-S.; et al. Validation of Susceptibility Loci for Vitiligo Identified by GWAS in the Chinese Han Population. Front. Genet. 2020, 11, 542275. [Google Scholar] [CrossRef]
- Montes, L.F.; Wilborn, W.H.; Hyde, B.M.; Vaughan, J.T.; Bennett, J.S. Vitiligo in a Quarter Horse Filly: Clinicopathologic, Ultrastructural, and Nutritional Study. J. Equine Vet. Sci. 2008, 28, 171–175. [Google Scholar] [CrossRef]
- Hofmanova, B.; Vostry, L.; Majzlik, I.; Vostra-Vydrova, H. Characterization of greying, melanoma, and vitiligo quantitative inheritance in Old Kladruber horses. Czech J. Anim. Sci. 2015, 60, 443–451. [Google Scholar]
- Solé, M.; Valera, M.; Sánchez, M.J.; Azor, P.J.; Fernández, J. Drawbacks and consequences of selective strategies in the design of semen banks: Case study of the Pura Raza Español horse breed. Livest. Sci. 2019, 226, 93–98. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, I.; Misztal, I.; Johnson, D.L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score1. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet. Res. 2012, 94, 73–83. [Google Scholar] [CrossRef]
- Misztal, I.; Tsuruta, S.; Lourenco, D.; Masuda, Y.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family of Programs; University of Georgia: Athens, GA, USA, 2016. [Google Scholar]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; the AmiGO Hub; the Web Presence Working Group. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M.; Tooker, M.E.; O’Connell, J.R.; Cole, J.B.; Bickhart, D.M. Selecting sequence variants to improve genomic predictions for dairy cattle. Genet. Sel. Evol. 2017, 49, 32. [Google Scholar] [CrossRef]
- Encina, A.; Valera, M.; Ligero, M.; Rodriguez Sainz de los Terreros, A.; Sánchez-Guerrero, M.J. Characterisation of white facial markings in Pura Raza Española horses (a worldwide population genetic study). Ital. J. Anim. Sci. 2024, 23, 929–937. [Google Scholar] [CrossRef]
- Encina, A.; Ligero, M.; Sánchez-Guerrero, M.J.; Rodríguez-Sainz de los Terreros, A.; Bartolomé, E.; Valera, M. Phenotypic and Genetic Study of the Presence of Hair Whorls in Pura Raza Español Horses. Animals 2023, 13, 2943. [Google Scholar] [CrossRef]
- McCreery, M.Q.; Halliwill, K.D.; Chin, D.; Delrosario, R.; Hirst, G.; Vuong, P.; Jen, K.-Y.; Hewinson, J.; Adams, D.J.; Balmain, A. Evolution of metastasis revealed by mutational landscapes of chemically induced skin cancers. Nat. Med. 2015, 21, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Georgiou, M.; Atkinson, R.; Collin, J.; Al-Aama, J.; Nagaraja-Grellscheid, S.; Johnson, C.; Ali, R.; Armstrong, L.; Mozaffari-Jovin, S.; et al. Pre-mRNA Processing Factors and Retinitis Pigmentosa: RNA Splicing and Beyond. Front. Cell Dev. Biol. 2021, 9, 700276. [Google Scholar] [CrossRef]
- Loftus, S.K.; Baxter, L.L.; Cronin, J.C.; Fufa, T.D.; Program, N.C.S.; Pavan, W.J. Hypoxia-induced HIF1α targets in melanocytes reveal a molecular profile associated with poor melanoma prognosis. Pigment Cell Melanoma Res. 2017, 30, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Bosch, P.J.; Peek, S.L.; Smolikove, S.; Weiner, J.A. Akirin proteins in development and disease: Critical roles and mechanisms of action. Cell. Mol. Life Sci. 2020, 77, 4237–4254. [Google Scholar] [CrossRef] [PubMed]
- Briard, B.; Place, D.E.; Kanneganti, T.-D. DNA Sensing in the Innate Immune Response. Physiology 2020, 35, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Valdivia-Silva, J.; Ramírez Díaz, C. Melanocytes in vitiligo and melanoma: A lesson between autoimmunity and tumor immunity. Dermatol. Peru. 2013, 23, 155–162. [Google Scholar]
- Huang, Y.; Wang, Y.; Wang, Y.; Wang, N.; Duan, Q.; Wang, S.; Liu, M.; Bilal, M.A.; Zheng, Y. LPCAT1 Promotes Cutaneous Squamous Cell Carcinoma via EGFR-Mediated Protein Kinase B/p38MAPK Signaling Pathways. J. Investig. Dermatol. 2022, 142, 303–313.e9. [Google Scholar] [CrossRef] [PubMed]
- Choquet, H.; Jiang, C.; Yin, J.; Kim, Y.; Hoffmann, T.J.; Aslibekyan, S.; Auton, A.; Babalola, E.; Bell, R.K.; Bielenberg, J.; et al. Multi-ancestry genome-wide meta-analysis identifies novel basal cell carcinoma loci and shared genetic effects with squamous cell carcinoma. Commun. Biol. 2024, 7, 33. [Google Scholar] [CrossRef]
- Maloberti, T.; De Leo, A.; Coluccelli, S.; Sanza, V.; Gruppioni, E.; Altimari, A.; Comito, F.; Melotti, B.; Marchese, P.V.; Dika, E.; et al. Molecular Characterization of Advanced-Stage Melanomas in Clinical Practice Using a Laboratory-Developed Next-Generation Sequencing Panel. Diagnostics 2024, 14, 800. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Mengxuan, Z.; Ming, R.; Zixu, G.; Yong, Z.; Simin, Z.; Yang, Y.; Leqi, Q.; Kangjie, S.; Yanlin, L.; et al. TRIP13/FLNA Complex Promotes Tumor Progression and Is Associated with Unfavorable Outcomes in Melanoma. J. Oncol. 2022, 2022, 1419179. [Google Scholar] [CrossRef] [PubMed]
- Mason, L.D.; Chava, S.; Reddi, K.K.; Gupta, R. The BRD9/7 Inhibitor TP-472 Blocks Melanoma Tumor Growth by Suppressing ECM-Mediated Oncogenic Signaling and Inducing Apoptosis. Cancers 2021, 13, 5516. [Google Scholar] [CrossRef] [PubMed]
- Dreier, M.R.; de la Serna, I.L. SWI/SNF Chromatin Remodeling Enzymes in Melanoma. Epigenomes 2022, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Hazane, F.; Valenti, K.; Sauvaigo, S.; Peinnequin, A.; Mouret, C.; Favier, A.; Beani, J.-C. Ageing effects on the expression of cell defence genes after UVA irradiation in human male cutaneous fibroblasts using cDNA arrays. J. Photochem. Photobiol. B Biol. 2005, 79, 171–190. [Google Scholar] [CrossRef]
- Liu, J.; Rebecca, V.W.; Kossenkov, A.V.; Connelly, T.; Liu, Q.; Gutierrez, A.; Xiao, M.; Li, L.; Zhang, G.; Samarkina, A.; et al. Neural Crest-Like Stem Cell Transcriptome Analysis Identifies LPAR1 in Melanoma Progression and Therapy Resistance. Cancer Res. 2021, 81, 5230–5241. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Khin, P.P.; Lim, O.K.; Jun, H.-S. LPA/LPAR1 signaling induces PGAM1 expression via AKT/mTOR/HIF-1α pathway and increases aerobic glycolysis, contributing to keratinocyte proliferation. Life Sci. 2022, 311, 121201. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, R.; Lusi, C.F.; Mashayekh, S.; Nagar, A.; Subbarao, M.; Kane, G.I.; Wodzanowski, K.A.; Brown, A.R.; Okuda, K.; Monahan, A.; et al. Methotrexate suppresses psoriatic skin inflammation by inhibiting muropeptide transporter SLC46A2 activity. Immunity 2023, 56, 998–1012.e8. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.B.; Islam, S.U.; Lee, Y.S. PRP4 Promotes Skin Cancer by Inhibiting Production of Melanin, Blocking Influx of Extracellular Calcium, and Remodeling Cell Actin Cytoskeleton. Int. J. Mol. Sci. 2021, 22, 6992. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; De La Torre, R.; Barling, A.; Tsujikawa, T.; Hornick, N.; Hanifin, J.; Simpson, E.; Wang, Y.; Swanzey, E.; et al. Trim32 Deficiency Enhances Th2 Immunity and Predisposes to Features of Atopic Dermatitis. J. Investig. Dermatol. 2017, 137, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ono, C.; Yu, Z.; Kasahara, Y.; Kikuchi, Y.; Ishii, N.; Tomita, H. Fluorescently Activated Cell Sorting Followed by Microarray Profiling of Helper T Cell Subtypes from Human Peripheral Blood. PLoS ONE 2014, 9, e111405. [Google Scholar] [CrossRef]
- Pośpiech, E.; Kukla-Bartoszek, M.; Karłowska-Pik, J.; Zieliński, P.; Woźniak, A.; Boroń, M.; Dąbrowski, M.; Zubańska, M.; Jarosz, A.; Grzybowski, T.; et al. Exploring the possibility of predicting human head hair greying from DNA using whole-exome and targeted NGS data. BMC Genom. 2020, 21, 538. [Google Scholar] [CrossRef]
- Min, J.; Zaslavsky, A.; Fedele, G.; McLaughlin, S.K.; Reczek, E.E.; De Raedt, T.; Guney, I.; Strochlic, D.E.; MacConaill, L.E.; Beroukhim, R.; et al. An oncogene–tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-κB. Nat. Med. 2010, 16, 286–294. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Score | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Categories | Absent | * | Few | * | Some | * | Many | * | Widespread |
| Degrees | Absent | Slight (2 ≤ score ≥ 5) | Severe (score ≥ 6) | ||||||
| Area | N | Descriptive Statistics | Prevalence (%) | |||
|---|---|---|---|---|---|---|
| Mean (s.d.) | Range | Absent | Slight | Severe | ||
| Eyes | 56,641 | 1.16 (0.908) | 1–9 | 96.01 | 2.92 | 1.07 |
| Mouth | 20,425 | 1.69 (1.565) | 1–9 | 79.07 | 16.99 | 3.95 |
| Nostrils | 56,632 | 1.57 (1.475) | 1–9 | 82.47 | 13.60 | 3.93 |
| Parameter | Trait | ||
|---|---|---|---|
| VE | VM | VN | |
| (s.d.) | 0.13 (0.006) | 0.13 (0.006) | 0.61 (0.022) |
| (s.d.) | 0.69 (0.006) | 0.83 (0.006) | 1.55 (0.017) |
| (s.d.) | 0.17 (0.007) | 0.13 (0.006) | 0.28 (0.009) |
| rg (s.d.) | VE–VM 0.55 (0.028) | VE–VN 0.52 (0.023) | VM–VN 0.79 (0.017) |
| Effect | Levels | VE | VM | VN |
|---|---|---|---|---|
| Inbreeding | b | 0.54 (0.101) | 0.73 (0.107) | 1.45 (0.163) |
| Sex | 1 | 1.31 (0.089) | 1.28 (0.085) | 1.79 (0.179) |
| 2 | 1.32 (0.089) | 1.27 (0.085) | 1.86 (0.179) | |
| Age | 1 | 0.10 (0.008) | 0.15 (0.008) | 0.24 (0.012) |
| 2 | <0.00 (0.00) | <0.00 (0.00) | <0.00 (0.00) | |
| Coat colour | 1 | −0.10 (0.018) | 0.13 (0.019) | −0.07 (0.029) |
| 2 | −0.21 (0.017) | −0.13 (0.018) | −0.49 (0.027) | |
| 3 | −0.12 (0.020) | −0.11 (0.021) | −0.48 (0.033) | |
| 4 | <0.00 (0.00) | <0.00 (0.00) | <0.00 (0.00) | |
| Geographic area | 1 | −0.09 (0.019) | <−0.00 (0.020) | −0.12 (0.032) |
| 2 | −0.03 (0.022) | 0.06 (0.023) | −0.07 (0.037) | |
| 3 | 0.01 (0.021) | −0.03 (0.022) | −0.08 (0.035) | |
| 4 | <0.00 (0.00) | <0.00 (0.00) | <0.00 (0.00) |
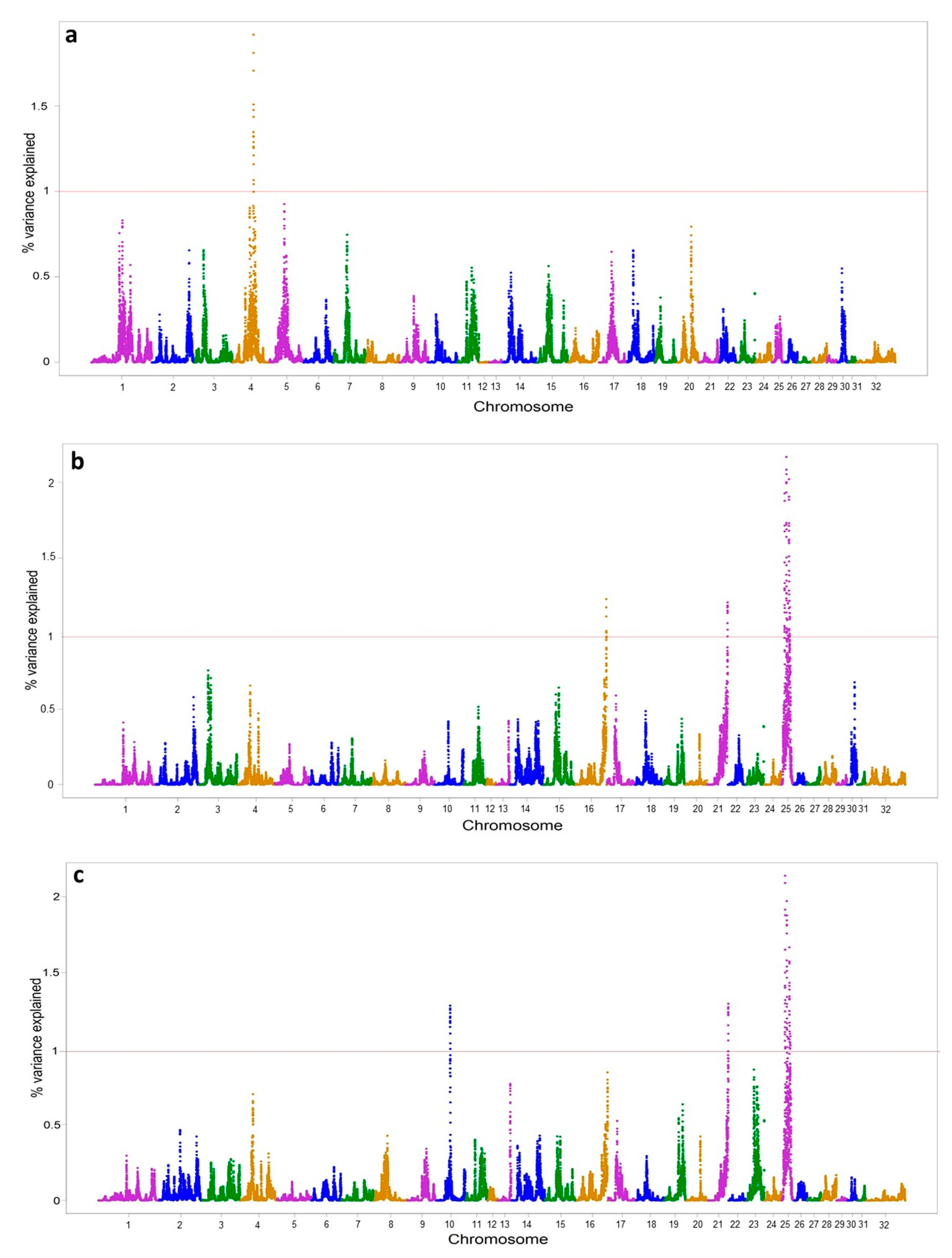
| Chr. | Genomic Region (pb) | v.e. (%) | Trait | Candidate Genes |
|---|---|---|---|---|
| 4 | 62,791,008–63,734,743 | 1.92 | VE | PDE1C; AVL9; KBTBD2; FKBP9; NT5C3A; RP9; BBS9 |
| 10 | 40,539,712–41,516,383 | 1.28 | VN | HTR1E; CGA; ZNF292; GJB7; SMIM8; C10H6orf163; CFAP206; RARS2; SLC35A1; ORC3; AKIRIN2 |
| 16 | 87,269,741–88,241,572 | 1.23 | VM | ARHGEF26; DHX36; GPR149 |
| 21 | 57,490,258–58,411,259 | 1.30 | VM and VN | IRX4; NDUFS6; MRPL36; LPCAT1; SLC6A3; CLPTM1L; TERT; SLC6A18; SLC6A19; SLC12A7; NKD2; TRIP13; BRD9 |
| 25 | 10,231,120–11,217,990 | 2.14 | VM and VN | TOPORSL; OR13F8; OR13D2H; OR13C2; OR13D2; OR13D2G; OR13C7O; OR13C8; OR13D1; NIPSNAP3A; NIPSNAP3B; ABCA1 |
| 25 | 15,753,928–16,753,198 | 2.17 | VM and VN | PALM2AKAP2; C25H9orf152; TXN; TXNDC8; SVEP1; MUSK; LPAR1; OR2K2; ECPAS |
| 25 | 17,727,314–18,708,795 | 1.09 | VM | SNX30; SLC46A2; SLC31A2; FKBP15; SLC31A1; CDC26; PRPF4; RNF183; WDR31; BSPRY; HDHD3; ALAD |
| 25 | 20,520,115–21,509,611 | 1.20 | VM and VN | PAPPA; ASTN2; TRIM32 |
| 25 | 23,519,210–24,493,448 | 2.02 | VM and VN | BRINP1 |
| 25 | 25,114,122–26,095,565 | 1.22 | VM and VN | C5; CNTRL; RAB14; GSN; STOM; DAB2IP; TTLL11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laseca, N.; Molina, A.; Perdomo-González, D.; Ziadi, C.; Azor, P.J.; Valera, M. Exploring the Genetic Landscape of Vitiligo in the Pura Raza Español Horse: A Genomic Perspective. Animals 2024, 14, 2420. https://doi.org/10.3390/ani14162420
Laseca N, Molina A, Perdomo-González D, Ziadi C, Azor PJ, Valera M. Exploring the Genetic Landscape of Vitiligo in the Pura Raza Español Horse: A Genomic Perspective. Animals. 2024; 14(16):2420. https://doi.org/10.3390/ani14162420
Chicago/Turabian StyleLaseca, Nora, Antonio Molina, Davinia Perdomo-González, Chiraz Ziadi, Pedro J. Azor, and Mercedes Valera. 2024. "Exploring the Genetic Landscape of Vitiligo in the Pura Raza Español Horse: A Genomic Perspective" Animals 14, no. 16: 2420. https://doi.org/10.3390/ani14162420
APA StyleLaseca, N., Molina, A., Perdomo-González, D., Ziadi, C., Azor, P. J., & Valera, M. (2024). Exploring the Genetic Landscape of Vitiligo in the Pura Raza Español Horse: A Genomic Perspective. Animals, 14(16), 2420. https://doi.org/10.3390/ani14162420