
Whole Mitochondrial Genome Sequencing and Phylogenetic Tree Construction for Procypris mera (Lin 1933)
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitochondrial Genome Sequencing and Assembly
2.3. Mitochondrial Genome Annotation and Analyses
2.4. Phlogenetic Analyses
3. Results
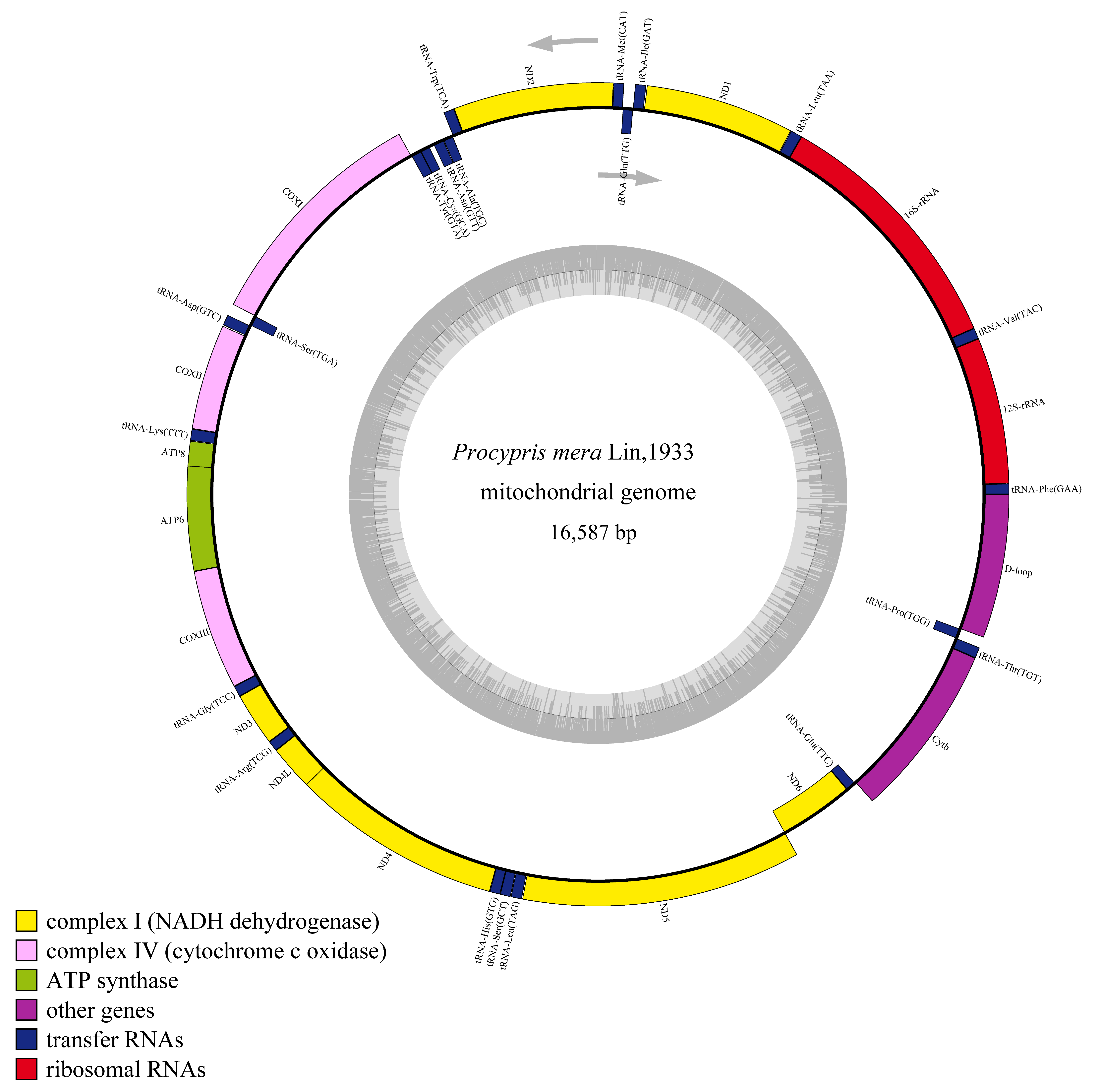
3.1. Mitochondrial Genome Analyses
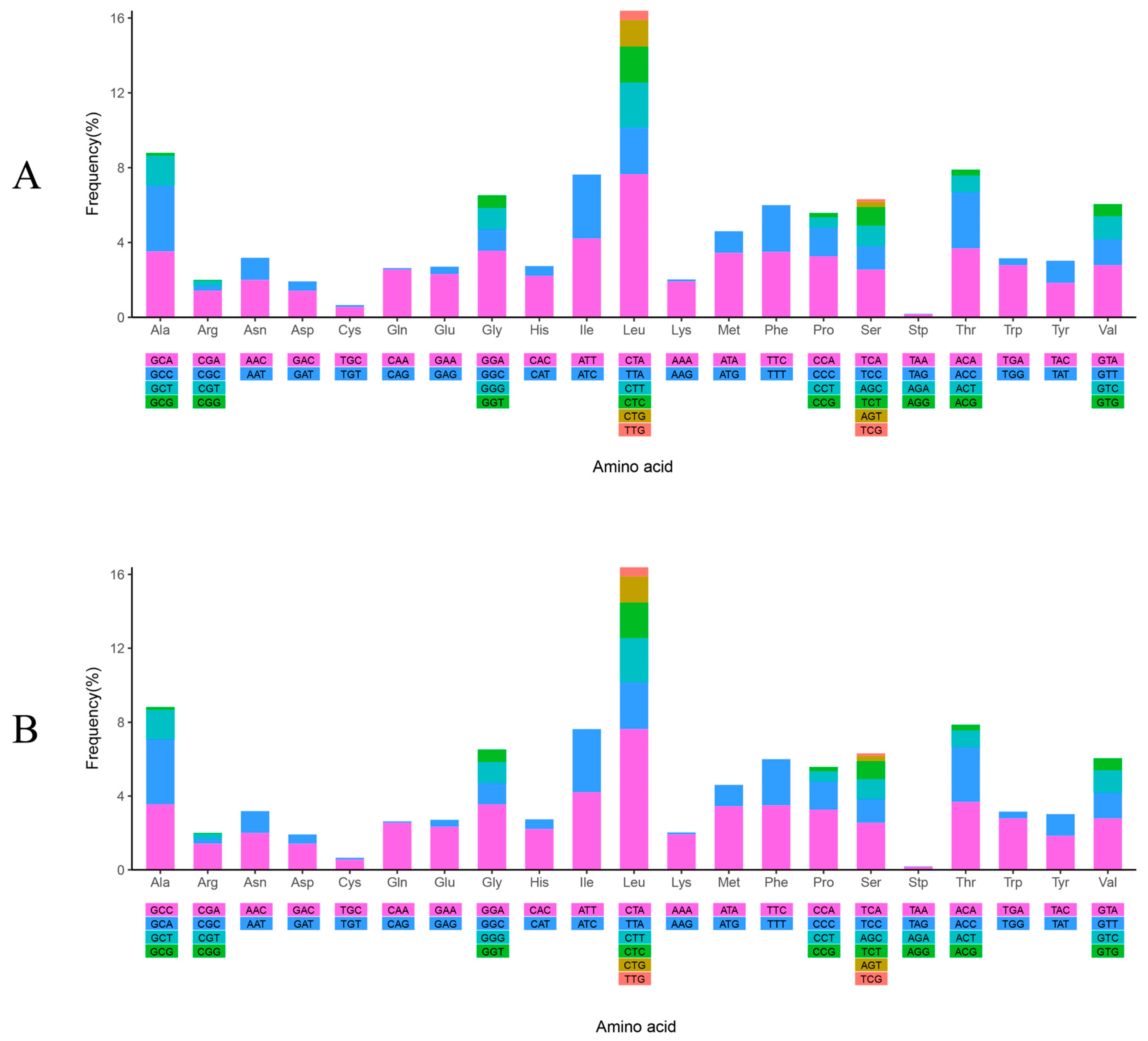
3.2. Protein Coding Gene Analyses
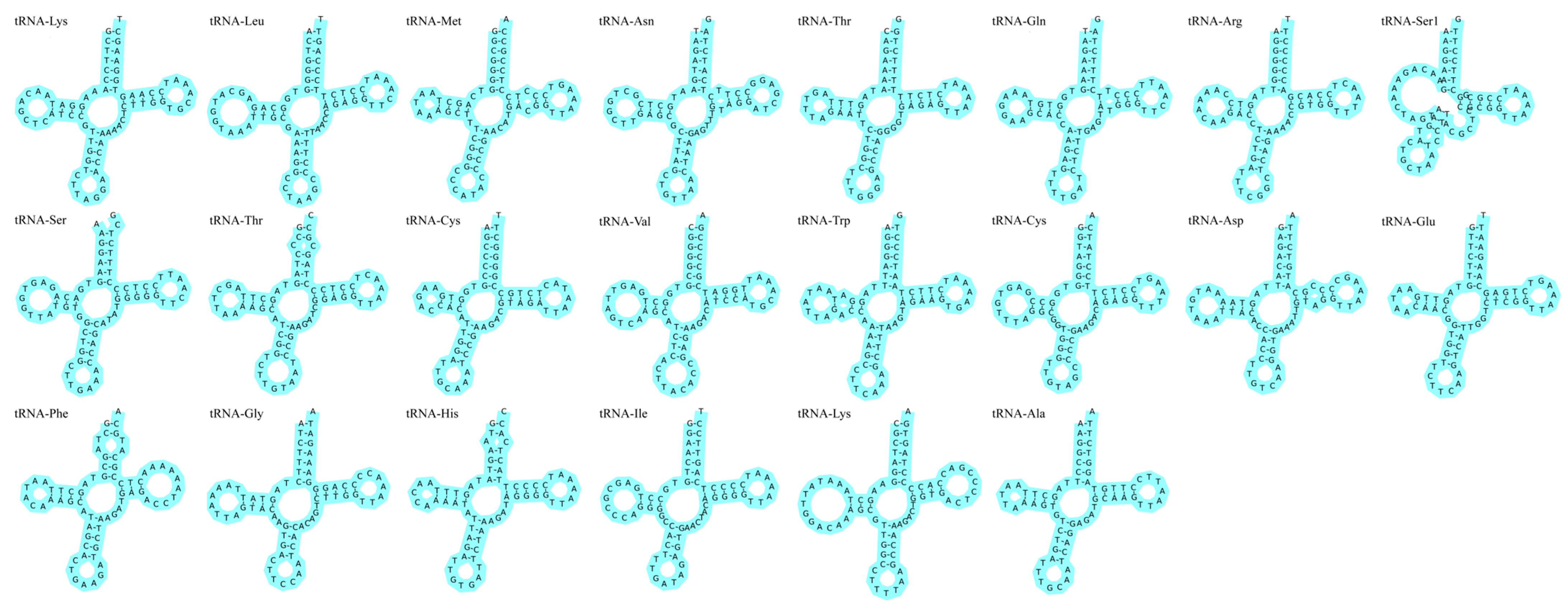
3.3. Ribosomal RNA, Transfer RNA Genes, and Non-Coding Regions Analyses
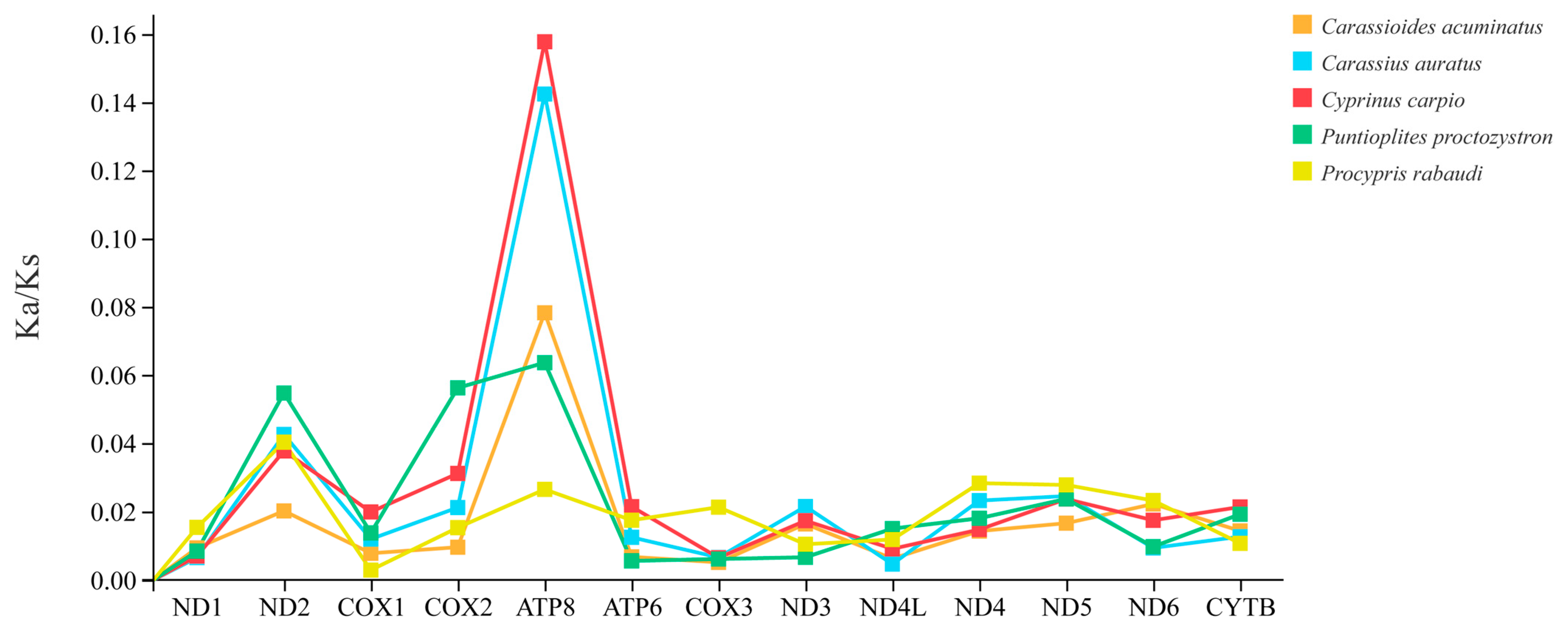
3.4. Ka/Ks Ratios Analyses
3.5. Phylogenetic Relationships
4. Discussion
4.1. Mitochondrial Differences between Individuals
4.2. Phylogenetic Relationships of Subfamily Cyprininae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guangxi Institute of Fishery Sciences; Z—Institute of Biophysics of Chinese Academy of Sciences. Freshwater Fishes of Guangxi, China; Guangxi People’s Press: Nanning, China, 1981. [Google Scholar]
- Yue, P.; Chen, Y. China Red Data Book of Endangered Animals Pisces; Science Press: Beijing, China, 1988. [Google Scholar]
- Han, Y.; He, A.; Lan, J.; Wu, W.; Li, Y. Phenotypic Traits and Fecundity Characteristics of Procypris mera. Jiangsu Agric. Sci. 2018, 46, 134–137. [Google Scholar]
- Han, Y.; He, A.; Lan, J.; Wu, W.; Li, Y.; Wang, D.; Lei, J.; Shi, J. Embryonic Development of Chinese Ink Carp. Fish. Sci. 2018, 37, 368–373. [Google Scholar]
- Lei, J.; Li, Y.; Li, Z.; Wang, D.; Shi, J.; Han, Y.; He, A.; Huang, B.; Wu, W. Relationship between Morphological Traits and Body Mass of Procypris mera. Chin. J. Fish. 2024, 37, 33–38. [Google Scholar]
- Grewe, P.; Hebert, P. Mitochondrial DNA Diversity among Broodstocks of the Lake Trout, Salvelinus Namaycush. Can. J. Fish. Aquat. Sci. 2011, 45, 2114–2122. [Google Scholar] [CrossRef]
- Vaughn, K.C.; Debonte, L.R.; Wilson, K.G.; Schaffer, G.W. Organelle Alteration as a Mechanism for Maternal Inheritance. Science 1980, 208, 196–198. [Google Scholar] [CrossRef]
- Meland, S.; Johansen, S.; Johansen, T.; Haugli, K.; Haugli, F. Rapid Disappearance of One Parental Mitochondrial Genotype after Isogamous Mating in the Myxomycete Physarum polycephalum. Curr. Genet. 1991, 19, 55–59. [Google Scholar] [CrossRef]
- Cantatore, P.; Saccone, C. Organization, Structure, and Evolution of Mammalian Mitochondrial Genes. In International Review of Cytology; Bourne, G.H., Jeon, K.W., Friedlander, M., Eds.; Academic Press: Cambridge, MA, USA, 1987; Volume 108, pp. 149–208. [Google Scholar]
- Zhang, K.; Liu, Y.; Yin, X.; Yuan, P.; Cao, P. Characterization of the Complete Mitochondrial Genome of Chinese Konosirus punctatus (Clupeiformes, Clupeidae) and Phylogenetic Studies of Clupeiformes. Mitochondrial DNA Part B 2020, 5, 3389–3391. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.; Liu, H. Phylogenetic Relationships of the Pseudogobionini Group (Teleostei: Cyprinidae) with Selection Pressure Analyses to Genes of Mitochondrial Genome. Fishes 2023, 8, 201. [Google Scholar] [CrossRef]
- Ficetola, G.F.; Miaud, C.; Pompanon, F.; Taberlet, P. Species Detection Using Environmental DNA from Water Samples. Biol. Lett. 2008, 4, 423–425. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic Annotation of Organellar Genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-Line: Integrating Search and Context for Analysis of Transfer RNA Genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A Program to Detect tRNA Genes in Metazoan Mitochondrial Nucleotide Sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.; Li, W.-H. The Codon Adaptation Index—A Measure of Directional Synonymous Codon Usage Bias, and Its Potential Applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A Parallel Tool for Constructing Multiple Protein-Coding DNA Alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit Incorporating Gamma-Series Methods and Sliding Window Strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Mu, H.; Chen, J.; Huang, W.; Huang, G.; Deng, M.; Hong, S.; Ai, P.; Gao, C.; Zhou, H. OmicShare Tools: A Zero-Code Interactive Online Platform for Biological Data Analysis and Visualization. iMeta 2024, e228. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.-Y.; Gao, F.; Jakovlić, I.; Lei, H.-P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.-T.; Zhang, D. Using phyloSuite for Molecular Phylogeny and Tree-Based Analyses. Imeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Oleinik, A.G.; Skurikhina, L.A.; Kukhlevsky, A.D.; Semenchenko, A.A. First Report of Three Complete Mitochondrial Genomes of the Long-Finned Charr Salvethymus svetovidovi Chereshnev et Skopetz, 1990 (Salmoniformes: Salmonidae) with Phylogenetic Consideration. Mitochondrial DNA B Resour. 2019, 4, 2464–2466. [Google Scholar] [CrossRef]
- Wahlund, S. Composition of Populations from the Perspective of the Theory of Heredity. Hereditas 1928, 11, 65–105. [Google Scholar] [CrossRef]
- Wang, Y. On the Classification, Distribution, Origin and Evolution of the Fishes Referred to the Subfamily Cyprininae of China, with Description of a New Species. ACTA Hydrobiol. Sin. 1979, 3, 419–438. [Google Scholar] [CrossRef]
- Chen, X.; Yue, P.; Lin, R. Major Groups within the Family Cyprinidae and Their Phylogenetic Relationships. Zool. Syst. 1984, 9, 424–440. [Google Scholar]
- Zhou, W. Phylogeny of the Subfamily Cyprinidae. ACTA Zootaxonomica Sin. 1989, 14, 247–256. [Google Scholar]
- van den Ende, C.; Puttick, M.N.; Urrutia, A.O.; Wills, M.A. Why Should We Compare Morphological and Molecular Disparity? Methods Ecol. Evol. 2023, 14, 2390–2410. [Google Scholar] [CrossRef]
- Yue, Z. The Monophyly of the Family Cyprinidae, the Barb Lineage, and the Subfamily Cyprinae and Their Relationships with Other Subfamilies Based on the D-Ring Region. Master’s Thesis, Yunnan University, Kunming, China, 2008. [Google Scholar]
- Luca, C.; Dudu, A.A.; Luca, A.; Dinischiotu, A.; Costache, M. Phylogenetic Relationships of Cyprinidae (Teleostei: Cypriniformes) Inferred from the Cox1 Gene Sequences; Studia Universitatis Vasile Goldis, Seria Stiintele Vietii (Life Sciences Series); Vasile Goldis University Press: Arad, Romania, 2008; Volume 18, pp. 175–180. [Google Scholar]
- Li, Q.; Li, W.; Han, C. Sequencing and Analysis of Complete Mitogenome of Cyprinus multitaeniate. J. Anhui Agric. Sci. 2024, 52, 108–111,120. [Google Scholar]
- Meng, Y.; Song, Y.; Xie, Q.; Cai, X.; Zhang, Q.; Dong, Y.; Li, F.; Xie, S.; Shen, Z. Mitochondrial Genome Sequencing and Structural Characteristic Analysis of Cyprinus acutidorsalis from Wanquan Estuary, Hainan. Mar. Sci. 2023, 47, 66–78. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | GenBank Accession Number | Length (bp) |
|---|---|---|---|
| Carassioides (Ōshima, 1926) | Carassioides acuminatus (J. Richardson, 1846) | KX602324.1 | 16,579 |
| Carassius (Nilsson, 1832) | Carassius auratus (Linnaeus, 1758) | EF483931.1 | 16,581 |
| Carassius carassius (Linnaeus, 1758) | AY714387.1 | 16,580 | |
| Carassius cuvieri (Temminck and Schlegel, 1846) | NC_010768.1 | 16,581 | |
| Carassius gibelio (Bloch, 1782) | GU138989.1 | 16,582 | |
| Carassius langsforfi (Temminck and Schlegel, 1846) | NC_002079.1 | 16,578 | |
| Cyprinus (Linnaeus, 1758) | Cyprinus megalophthalmus (Wu et al., 1963) | KR869143.1 | 16,580 |
| Cyprinus carpio (Linnaeus, 1758) | NC_001606.1 | 16,575 | |
| Cyprinus multitaeniata (Pellegrin and Chevey, 1936) | KR869145.1 | 16,580 | |
| Cyprinus pellegrini (Tchang, 1933) | MN718675.1 | 16,583 | |
| Cyprinus rubrofuscus (Lacepede, 1803) | MW969691.1 | 16,582 | |
| Cyprinus acutidorsalis (Chen and Hwang, 1977) | KR869144.1 | 16,580 | |
| Procypris | Procypris mera2019 (Lin, 1933) | MN841761.1 | 16,579 |
| Procypris mera2012 (Lin, 1933) | JX316027.1 | 16,586 | |
| Procypris meraWLY1 (Lin, 1933) | MN229744 | 16,587 | |
| Procypris meraWLY2 (Lin, 1933) | MN229745 | 16,587 | |
| Procypris meraWLY3 (Lin, 1933) | MN229746 | 16,587 | |
| Procypris rabaudi (Tchang, 1930) | EU082030.1 | 16,595 | |
| Puntioplites (H. M. Smith, 1929) | Puntioplites proctozystron (Bleeker, 1865) | AP011247.1 | 16,584 |
| Misgurnus (Lacépède, 1803) | Misgurnus anguillicaudatus (Cantor, 1842) | MF579257.1 | 16,647 |
| Length (bp) | Reads | Base (G) | Q20 | Q30 | GC (%) | |
|---|---|---|---|---|---|---|
| WYL1 | 150 | 4,596,214 | 1.38 | 96.47 | 91.95 | 38.16 |
| WYL2 | 150 | 4,288,544 | 1.29 | 96.20 | 91.45 | 38.16 |
| WYL3 | 150 | 3,490,319 | 1.05 | 96.23 | 91.47 | 38.41 |
| A (bp) | T (bp) | G (bp) | C (bp) | Length (bp) | GC (%) | GC-skew (%) | AT-skew (%) | |
|---|---|---|---|---|---|---|---|---|
| WYL1 | 5304 | 4133 | 2633 | 4517 | 16,587 | 43.1 | −0.2635 | 0.1241 |
| WYL2 | 5303 | 4133 | 2634 | 4517 | 16,587 | 43.1 | −0.2633 | 0.124 |
| WYL3 | 5303 | 4133 | 2634 | 4517 | 16,587 | 43.1 | −0.2633 | 0.124 |
| Gene /Element | Location | Length (bp) | Strand | Initiation Codon | Termination Codon | Intergenic Nucleotide (bp) | Gene /Element | Location | Length (bp) | Strand | Initiation Codon | Termination Codon | Intergenic Nucleotide (bp) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tRNA-Phe | 1–69 | 69 | H | 0 | tRNA-Lys | 7872–7947 | 76 | H | 1 | ||||
| 12S-rRNA | 70–1024 | 955 | H | 0 | ATP8 | 7949–8113 | 165 | H | ATG | TAG | −7 | ||
| tRNA-Val | 1025–1096 | 72 | H | 0 | ATP6 | 8107–8790 | 684 | H | ATG | TAA | −1 | ||
| 16S-rRNA | 1097–2779 | 1683 | H | 0 | COXIII | 8790–9574 | 785 | H | ATG | TA | 0 | ||
| tRNA-Leu | 2780–2855 | 76 | H | 1 | tRNA-Gly | 9575–9646 | 72 | H | 0 | ||||
| ND1 | 2857–3831 | 975 | H | ATG | TAA | 4 | ND3 | 9647–9995 | 349 | H | ATG | T | 0 |
| tRNA-Ile | 3836–3907 | 72 | H | −2 | tRNA-Arg | 9996–10,065 | 70 | H | 0 | ||||
| tRNA-Gln | 3906–3976 | 71 | L | 1 | ND4L | 10,066–10,362 | 297 | H | ATG | TAA | −7 | ||
| tRNA-Met | 3978–4046 | 69 | H | 0 | ND4 | 10,356–11,736 | 1381 | H | ATG | T | 0 | ||
| ND2 | 4047–5091 | 1045 | H | ATG | T | 0 | tRNA-His | 11,737–11,805 | 69 | H | 0 | ||
| tRNA-Trp | 5092–5162 | 71 | H | 2 | tRNA-Ser | 11,806–11,874 | 69 | H | 1 | ||||
| tRNA-Ala | 5165–5233 | 69 | L | 1 | tRNA-Leu | 11,876–11,948 | 73 | H | 3 | ||||
| tRNA-Asn | 5235–5307 | 73 | L | 33 | ND5 | 11,952–13,775 | 1824 | H | ATG | TAA | −4 | ||
| tRNA-Cys | 5341–5407 | 67 | L | −1 | ND6 | 13,772–14,293 | 522 | L | ATG | TAA | 0 | ||
| tRNA-Tyr | 5407–5477 | 71 | L | 1 | tRNA-Glu | 14,294–14,362 | 69 | L | 5 | ||||
| COXI | 5479–7029 | 1551 | H | GTG | TAA | 0 | Cytb | 14,368–15,508 | 1141 | H | ATG | T | 0 |
| tRNA-Ser | 7030–7100 | 71 | L | 3 | tRNA-Thr | 15,509–15,580 | 72 | H | −1 | ||||
| tRNA-Asp | 7104–7175 | 72 | H | 5 | tRNA-Pro | 15,580–15,649 | 70 | L | 0 | ||||
| COXII | 7181–7871 | 691 | H | ATG | T | 0 | D-loop | 15,650–16,587 | 938 | H | 0 |
| Position | WYL2 | WYL1 | Gene Type | Gene | Codon | Amino Acid | Mutation Type |
|---|---|---|---|---|---|---|---|
| 1296 | A | G | rRNA | 16S-rRNA | SNP | ||
| 7354 | C | T | CDS | COX2 | TCC > TCT | Ser > Ser | SNP |
| 8502 | A | G | CDS | ATP6 | GAA > GAG | Glu > Glu | SNP |
| 12,774 | G | A | CDS | ND5 | GCC > ACC | Ala > Thr | SNP |
| 14,503 | T | C | CDS | CYTB | TTA > CTA | Leu > Leu | SNP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Han, Y.; Li, Y.; Wu, W.; Lei, J.; Wang, D.; Lin, Y.; Wang, X. Whole Mitochondrial Genome Sequencing and Phylogenetic Tree Construction for Procypris mera (Lin 1933). Animals 2024, 14, 2672. https://doi.org/10.3390/ani14182672
Li Z, Han Y, Li Y, Wu W, Lei J, Wang D, Lin Y, Wang X. Whole Mitochondrial Genome Sequencing and Phylogenetic Tree Construction for Procypris mera (Lin 1933). Animals. 2024; 14(18):2672. https://doi.org/10.3390/ani14182672
Chicago/Turabian StyleLi, Zhe, Yaoquan Han, Yusen Li, Weijun Wu, Jianjun Lei, Dapeng Wang, Yong Lin, and Xiaoqing Wang. 2024. "Whole Mitochondrial Genome Sequencing and Phylogenetic Tree Construction for Procypris mera (Lin 1933)" Animals 14, no. 18: 2672. https://doi.org/10.3390/ani14182672