
Genetic Characterization of the Co-Invasive Rodent Parasite Heterakis spumosa (Nematoda, Heterakidae)

 , , ,
, , ,  ,
,
Abstract
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction
2.3. DNA Amplification and Sequencing
2.4. Sequence Alignment and Phylogenetic
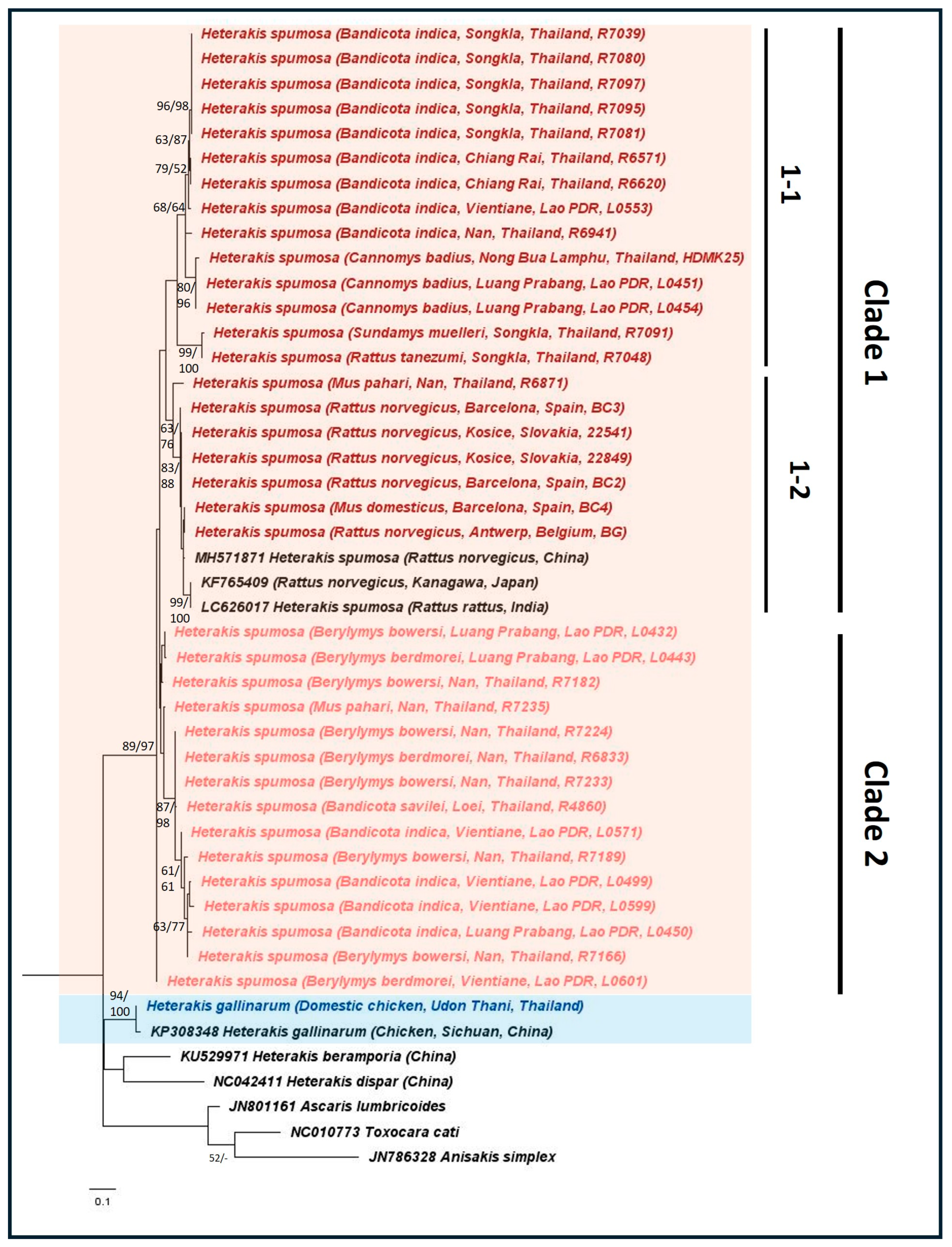
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milazzo, C.; Goüy de Bellocq, J.; Cagnin, M.; Casanova, J.C.; Di Bella, C.; Feliu, C.; Fons, R.; Morand, S.; Santalla, F. Helminths and ectoparasites of Rattus rattus and Mus musculus from Sicily, Italy. Comp. Parasitol. 2003, 70, 199–204. [Google Scholar] [CrossRef]
- Milazzo, C.; Ribas, A.; Casanova, J.C.; Cagnin, M.; Geraci, F.; Di Bella, C. Helminths of the brown rat (Rattus norvegicus) (Berkenhout, 1769) in the City of Palermo, Italy. Helminthologia 2010, 47, 238–240. [Google Scholar] [CrossRef]
- Zaleśny, G.; Hildebrand, J.; Popiołek, M. Molecular Identification of Heterakis spumosa Schneider, 1866 (Nematoda: Ascaridida: Heterakidae) with comparative analysis of its occurrence in two mice species. Ann. Zool. 2010, 60, 647–655. [Google Scholar] [CrossRef]
- Ribas, A.; de Bellocq, J.G.; Ros, A.; Ndiaye, P.I.; Miquel, J. Morphometrical and genetic comparison of two nematode species: H. spumosa and H. dahomensis (Nematoda, Heterakidae). Acta Parasitol. 2013, 58, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Šnábel, V.; Utsuki, D.; Kato, T.; Sunaga, F.; Ooi, H.K.; Gambetta, B.; Taira, K. Molecular identification of Heterakis spumosa obtained from brown rats (Rattus norvegicus) in Japan and its infectivity in experimental mice. Parasitol. Res. 2014, 113, 3449–3455. [Google Scholar] [CrossRef] [PubMed]
- Julius, R.S.; Schwan, E.V.; Chimimba, C.T. Helminth composition and prevalence of indigenous and invasive synanthropic murid rodents in urban areas of Gauteng Province, South Africa. J. Helminthol. 2017, 92, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Hancke, D.; Suarez, O.V. Structure of parasite communities in urban environments: The case of helminths in synanthropic rodents. Folia Parasitol. 2018, 65, 009. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhu, J.Y.; Jian, K.L.; Wang, B.J.; Peng, X.R.; Yang, G.Y.; Wang, T.; Zhong, Z.J.; Peng, K.Y. Absence of population genetic structure in Heterakis gallinarum of chicken from Sichuan, inferred from mitochondrial cytochrome C oxidase subunit I gene. Mitochondrial DNA 2016, 29 Pt A, 629–634. [Google Scholar] [CrossRef]
- Wang, Y.; Feijó, A.; Cheng, J.; Xia, L.; Wen, Z.; Ge, D.; Sun, J.; Lu, L.; Li, S.; Yang, Q. Ring Distribution patterns—Diversification or speciation? Comparative phylogeography of two small mammals in the mountains surrounding the Sichuan Basin. Mol. Ecol. 2021, 30, 2641–2658. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, L.N.; Sabuni, C.; Šumbera, R.; Aghová, T.; Lišková, E.; Leirs, H.; Baird, S.J.E.; Goüy de Bellocq, J.; Bryja, J. Biogeographical importance of the Livingstone mountains in southern Tanzania: Comparative genetic structure of small non-volant Mammals. Front. Ecol. Evol. 2022, 9, 742851. [Google Scholar] [CrossRef]
- Brunke, J.; Radespiel, U.; Russo, I.; Bruford, M.; Goossens, B. Messing about on the river: The role of geographic barriers in shaping the genetic structure of bornean small mammals in a fragmented landscape. Conserv. Genet. 2019, 20, 691–704. [Google Scholar] [CrossRef]
- Ribas, A.; Chaisiri, K.; Morand, S.; Hugot, J.P.; Haukisalmi, V.; Henttonen, H. Isolation of helminths from rodents. In Protocols for Field and Laboratory Rodent Studies; Herbreteau, V., Jittapalapong, S., Rerkamnuaychoke, W., Chaval, Y., Cosson, J.F., Morand, S., Eds.; Kasetsart University Press: Bangkok, Thailand, 2011; pp. 32–35. [Google Scholar]
- Hall, T.A. BioEdit: A User friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002, 2, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree, version 1.3.1; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2009. [Google Scholar]
- Morand, S.; Bordes, F.; Hsuan-Wien, C.; Julien, C.; Cosson, J.; Maxime, G.; Czirják, G.; Greenwood, A.; Latinne, A.; Michaux, J.; et al. Global parasite and Rattus rodent invasions: The Consequences for rodent-borne diseases. Integr. Zool. 2015, 10, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Poulin, R.; Krasnov, B.R.; Morand, S. Patterns of Host Specificity in parasites exploiting small mammals. In Micromammals and Macroparasites: From Evolutionary Ecology to Management; Springer: Tokyo, Japan, 2006; pp. 233–256. ISBN 9784431360254. [Google Scholar]
- Epps, C.W.; Keyghobadi, N. Landscape genetics in a changing world: Disentangling historical and contemporary influences and inferring change. Mol. Ecol. 2015, 24, 6021–6040. [Google Scholar] [CrossRef] [PubMed]
- Jueco, N.L.; Zabala, Z.R. The nematodes of Rattus norvegicus and Rattus rattus mindanensis. Philipp. J. Vet. Med. 1990, 27, 39–46. [Google Scholar]
- Mohd Zain, S.N.; Behnke, J.M.; Lewis, J.W. Helminth communities from two urban rat populations in Kuala Lumpur, Malaysia. Parasites Vectors 2012, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Dewi, K.; Purwaningsih, E. Helminth parasites on rats in rubber plantation in Bogorejo Village, Gedongtataan Subdistrict, Pesawaran Regency, Lampung and their zoonotic review. Zoo Indones. 2013, 22, 1–7. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Genetic Marker | Primers Designed | Annealing Temperature (°C) | PCR Amplicons Size (bp) | Accession No. of DNA Sequences Used for Primer Design |
|---|---|---|---|---|
| mtCOI | Hs-COX1_Fwd | 50 | 386 | MH571871, MH571870, NC_029838, NC_029839, OR177661, KY368775, KT900946, KT764937 |
| 5′GAT TTT GCC TGC TTT TGG TAT 3′ | ||||
| Hs-COX1_REV | ||||
| 5′CTC ACC ACA TAA TAA GTA TCA 3′ | ||||
| Nuclear rRNA ITS1 | ITS1-Heterakis-F | 60 | 563 | JX845277, JX845278, MF319969, MH215352, EF464551, EF464554, EF464553, KP776414, KU575094 |
| 5′GAG CCR CGT AGT GTA CTA CAA 3′ | ||||
| ITS1-Heterakis-R | ||||
| 5′TCG ACC CTC AGC CAG ACG TG 3′ |
| Species/Genetic Distance | H. spumosa | H. dispar | H. gallinarum | H. beramporia | ||
|---|---|---|---|---|---|---|
| Clade 1 | Clade 2 | |||||
| H. spumosa | Clade 1 | 7% | ||||
| Clade 2 | 8.20% | 4% | ||||
| H. dispar | 14.30% | 13.10% | ||||
| H. gallinarum | 14.10% | 13.30% | 13.60% | |||
| H. beramporia | 14.00% | 14.20% | 14.40% | 12.90% | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poonlaphdecha, S.; Ribas, A.; Chaisiri, K.; Morand, S.; Chan, A.H.E.; Thaenkham, U. Genetic Characterization of the Co-Invasive Rodent Parasite Heterakis spumosa (Nematoda, Heterakidae). Animals 2024, 14, 2674. https://doi.org/10.3390/ani14182674
Poonlaphdecha S, Ribas A, Chaisiri K, Morand S, Chan AHE, Thaenkham U. Genetic Characterization of the Co-Invasive Rodent Parasite Heterakis spumosa (Nematoda, Heterakidae). Animals. 2024; 14(18):2674. https://doi.org/10.3390/ani14182674
Chicago/Turabian StylePoonlaphdecha, Srisupaph, Alexis Ribas, Kittipong Chaisiri, Serge Morand, Abigail Hui En Chan, and Urusa Thaenkham. 2024. "Genetic Characterization of the Co-Invasive Rodent Parasite Heterakis spumosa (Nematoda, Heterakidae)" Animals 14, no. 18: 2674. https://doi.org/10.3390/ani14182674
APA StylePoonlaphdecha, S., Ribas, A., Chaisiri, K., Morand, S., Chan, A. H. E., & Thaenkham, U. (2024). Genetic Characterization of the Co-Invasive Rodent Parasite Heterakis spumosa (Nematoda, Heterakidae). Animals, 14(18), 2674. https://doi.org/10.3390/ani14182674