
Combining Transcriptomics and Proteomics to Screen Candidate Genes Related to Bovine Birth Weight
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Placenta Sample Collection
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. Quality Control, Annotation, and Differential Expression Analysis
2.4. Protein Extraction and Digestion
2.5. iTRAQ Labeling and HPLC Fractionation
2.6. Liquid Chromatography Tandem–Mass Spectrometry(LC-MS/MS) and Data Analysis
2.7. Bioinformatics Analyses
2.8. Quantitative Real-Time PCR
2.9. Western Blot
2.10. Statistical Analysis
3. Results
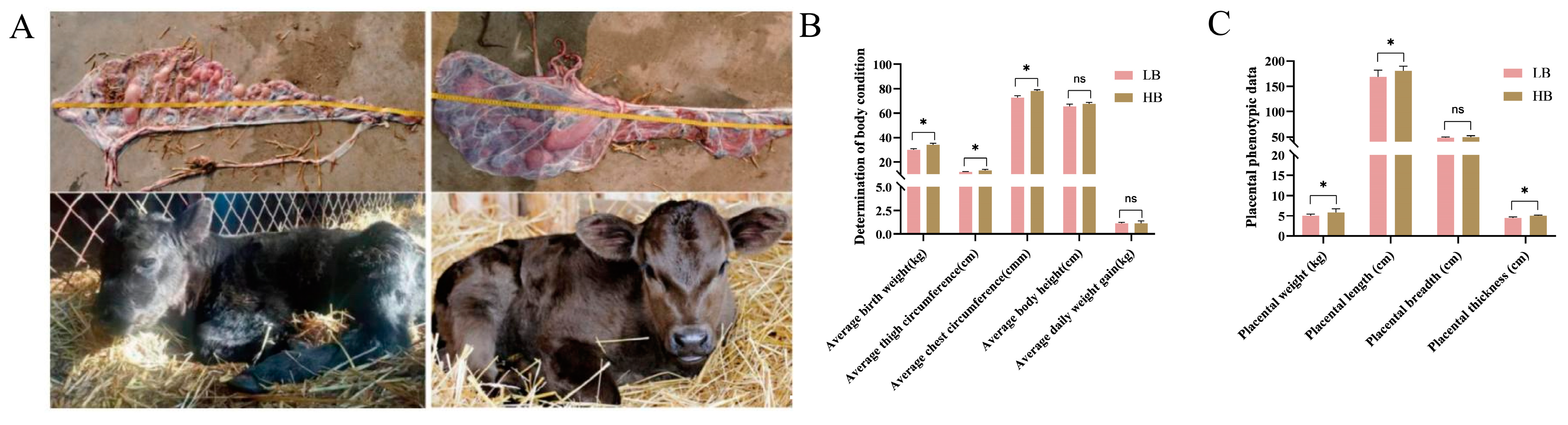
3.1. Characterization of Calves
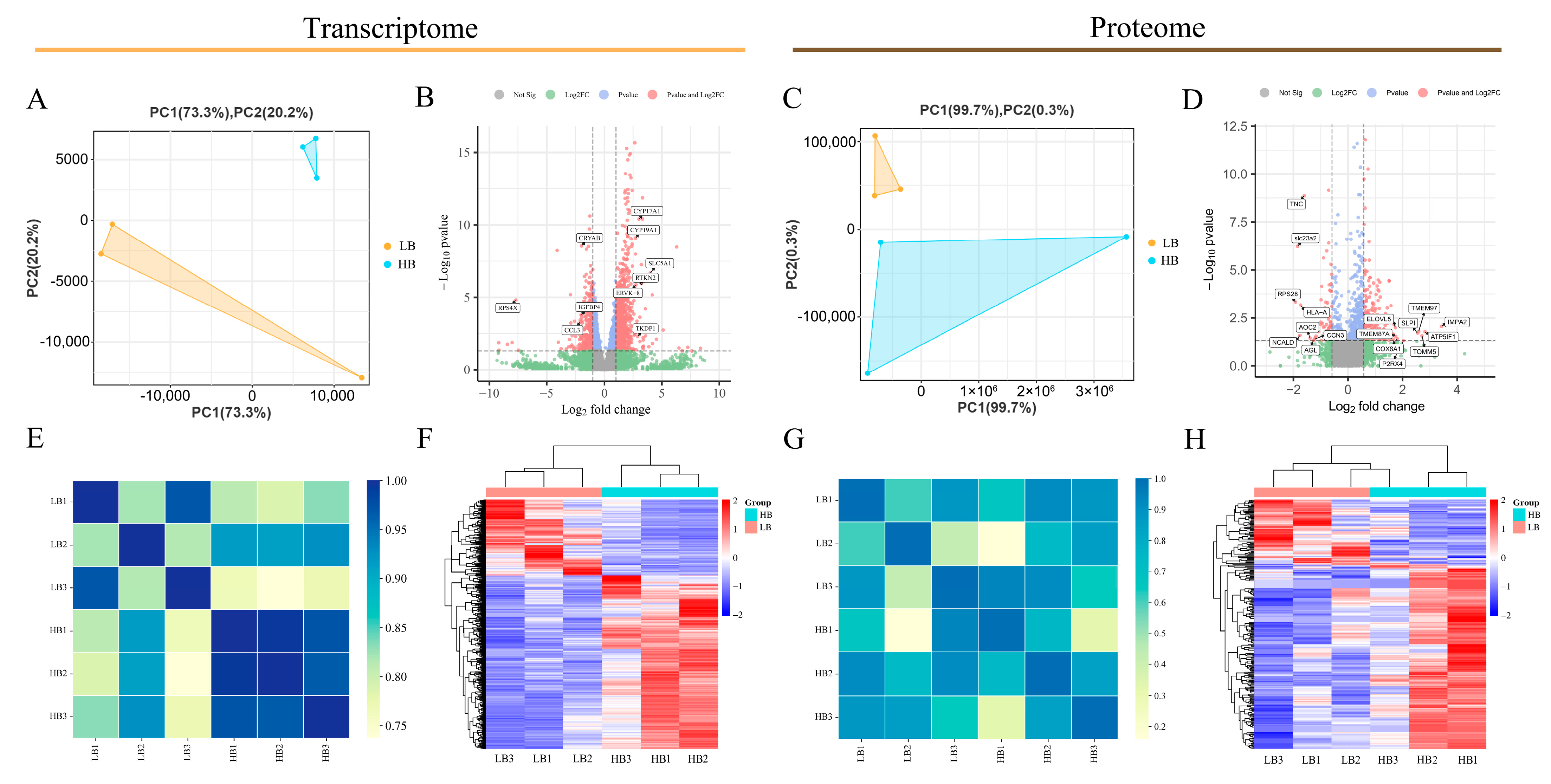
3.2. Overall Transcriptomic and Proteomic Analysis Statistics
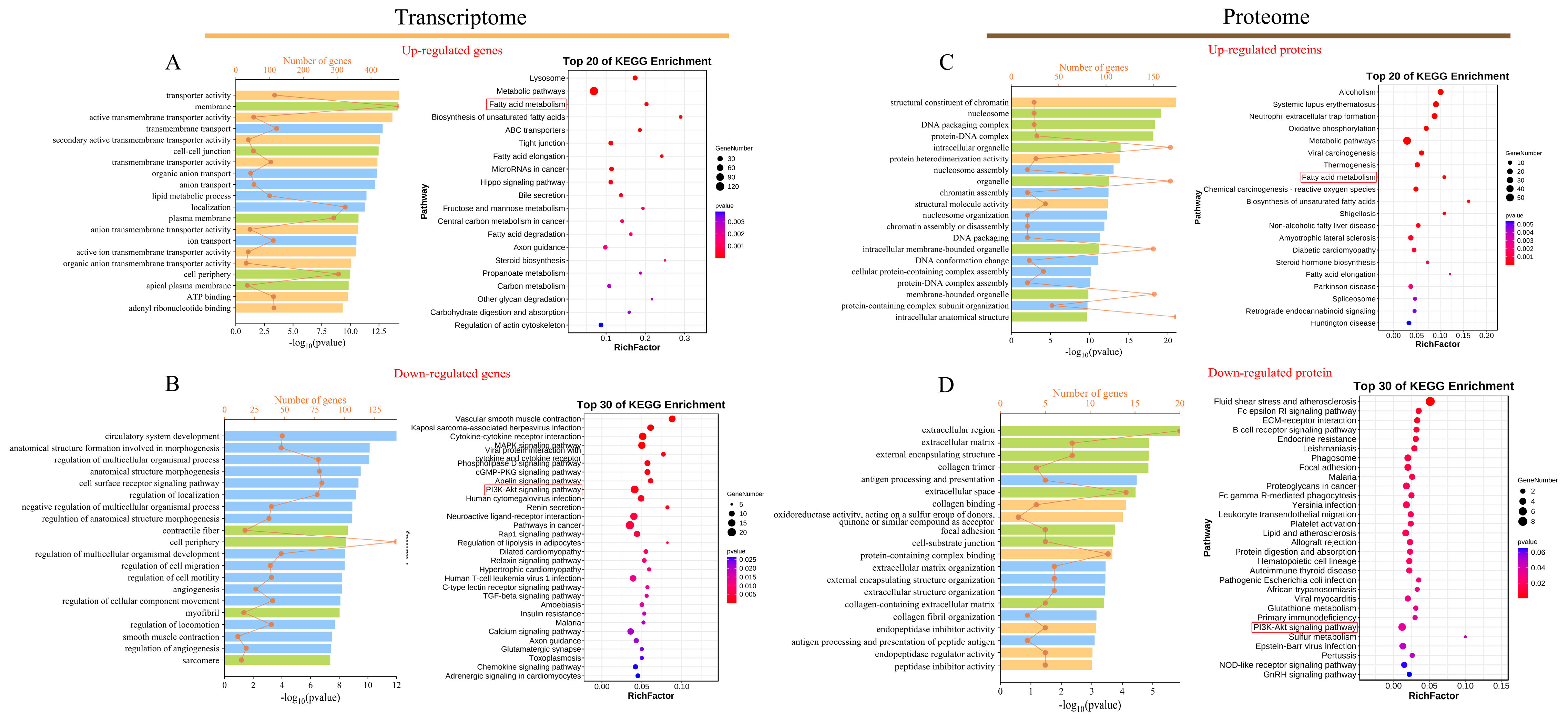
3.3. Functional Analysis of DEGs
3.4. Functional Analysis of DEPs
3.5. Integrative Analysis of Proteomics and Transcriptomics
3.6. Protein–Protein Interaction Analysis
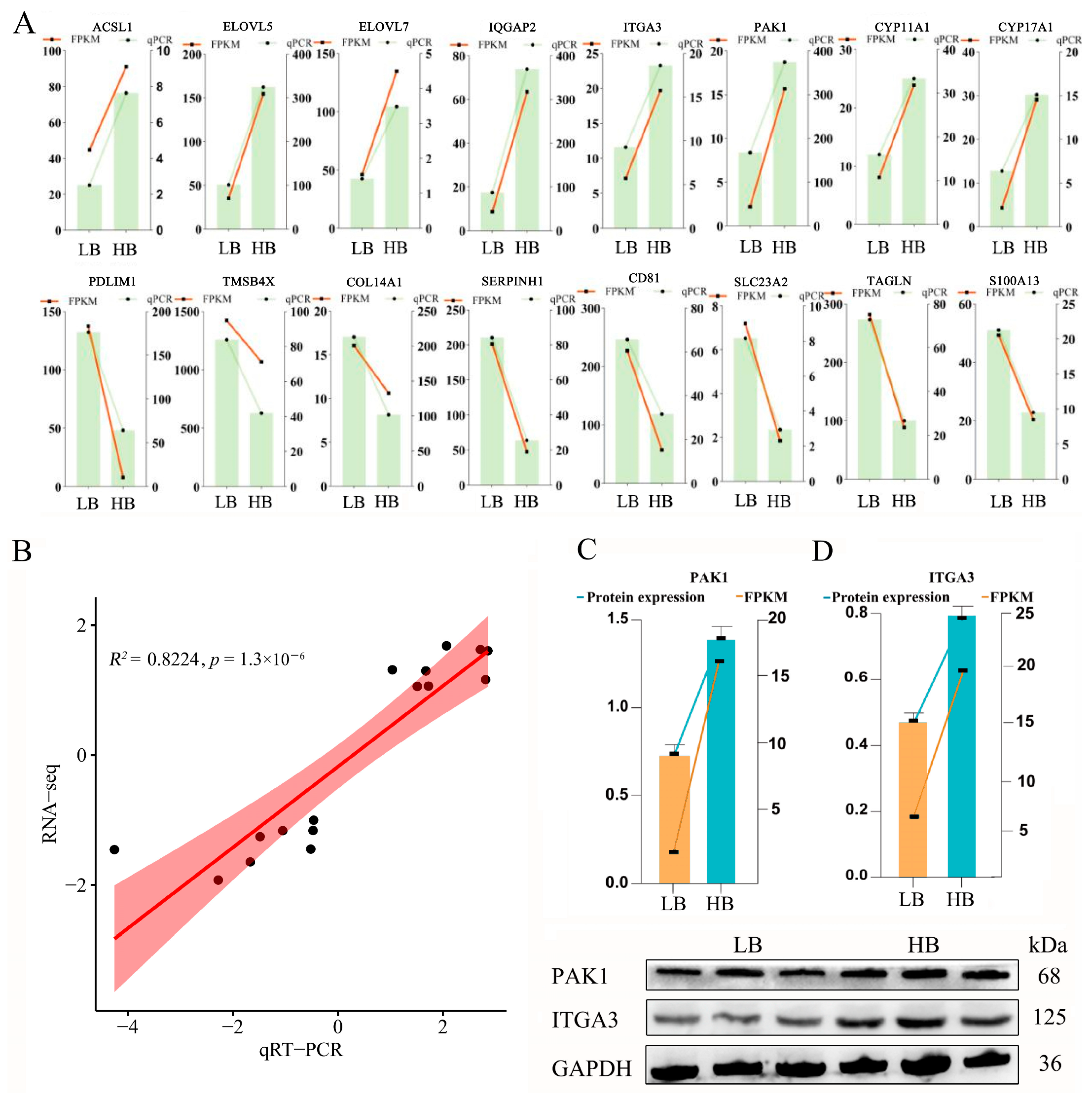
3.7. qRT-PCR and Western Blot Validation of Candidate DEGs/DEPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McNeill, S.H. Inclusion of red meat in healthful dietary patterns. Meat Sci. 2014, 98, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, P.L. Review: An overview of beef production from pasture and feedlot globally, as demand for beef and the need for sustainable practices increase. Animal 2021, 15 (Suppl. 1), 100295. [Google Scholar] [CrossRef] [PubMed]
- Iida, F.; Saitou, K.; Kawamura, T.; Yamaguchi, S.; Nishimura, T. Effect of fat content on sensory characteristics of marbled beef from Japanese Black steers. Anim. Sci. J. 2015, 86, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Daley, C.A.; Abbott, A.; Doyle, P.S.; Nader, G.A.; Larson, S. A review of fatty acid profiles and antioxidant content in grass-fed and grain-fed beef. Nutr. J. 2010, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Van Elswyk, M.E.; McNeill, S.H. Impact of grass/forage feeding versus grain finishing on beef nutrients and sensory quality: The U.S. experience. Meat Sci. 2014, 96, 535–540. [Google Scholar] [CrossRef]
- Gotoh, T.; Albrecht, E.; Teuscher, F.; Kawabata, K.; Sakashita, K.; Iwamoto, H.; Wegner, J. Differences in muscle and fat accretion in Japanese Black and European cattle. Meat Sci. 2009, 82, 300–308. [Google Scholar] [CrossRef]
- Wang, J.; Fan, T.; Du, Z.; Xu, L.; Chen, Y.; Zhang, L.; Gao, H.; Li, J.; Ma, Y.; Gao, X. Genome-Wide Association Analysis Identifies the PMEL Gene Affecting Coat Color and Birth Weight in Simmental × Holstein. Animals 2023, 13, 3821. [Google Scholar] [CrossRef]
- te Velde, S.J.; Twisk, J.W.; van Mechelen, W.; Kemper, H.C. Birth weight and musculoskeletal health in 36-year-old men and women: Results from the Amsterdam Growth and Health Longitudinal Study. Osteoporos. Int. 2004, 15, 382–388. [Google Scholar] [CrossRef]
- Heng-Wei, Y.; Raza, S.H.A.; Almohaimeed, H.M.; Alzahrani, S.S.; Alkhalifah, S.M.; Yasir, B.A.L.; Yasir, B.A.L.; Zan, L. The body weight heritability and the effect of non-genetic factors on the body measurement traits in Qinchuan cattle. Anim. Biotechnol. 2023, 34, 4387–4393. [Google Scholar] [CrossRef]
- Hickson, R.E.; Morris, S.T.; Kenyon, P.R.; Lopez-Villalobos, N. Dystocia in beef heifers: A review of genetic and nutritional influences. N. Z. Vet. J. 2006, 54, 256–264. [Google Scholar] [CrossRef]
- Yáñez-Muñoz, A.; Rosete-Fernández, J.V.; González-Orozco, T.A.; Ríos-Utrera, Á. Phenotypic evaluation of pre-weaning growth traits of Akaushi- (Wagyu), Angus- and Brahman-sired calves in tropical conditions. Trop. Anim. Health Prod. 2023, 55, 210. [Google Scholar] [CrossRef] [PubMed]
- da Silveira, D.D.; Schmidt, P.I.; Campos, G.S.; de Vargas, L.; de Souza, F.R.P.; Roso, V.M.; Boligon, A.A. Genetic analysis of growth, visual scores, height, and carcass traits in Nelore cattle. Anim. Sci. J. 2021, 92, e13611. [Google Scholar] [CrossRef] [PubMed]
- Kassahun, D.; Taye, M.; Kebede, D.; Tilahun, M.; Tesfa, A.; Bitew, A.; Kebede, A.; Meseret, M.; Lakew, E.; Bimrow, T.; et al. Phenotypic and genetic parameter estimates for early growth, growth rate and growth efficiency-related traits of Fogera cattle in Ethiopia. Vet. Med. Sci. 2022, 8, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Mallia, T.; Grech, A.; Hili, A.; Calleja-Agius, J.; Pace, N.P. Genetic determinants of low birth weight. Minerva Ginecol. 2017, 69, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Liu, Y.; Ji, B.; Fang, Y.; Xie, Y.; Sakurai, R.; Wang, J.; Zhang, Z.; Wang, Y.; Wang, X.; et al. Evidence for Wnt signaling’s central involvement in perinatal nicotine exposure-induced offspring lung pathology and its modulation by electroacupuncture. Biomed. Pharmacother. 2023, 168, 115824. [Google Scholar] [CrossRef]
- Morsing, E.; Liuba, P.; Fellman, V.; Marsal, K.; Brodszki, J. Cardiovascular function in children born very preterm after intrauterine growth restriction with severely abnormal umbilical artery blood flow. Eur. J. Prev. Cardiol. 2014, 21, 1257–1266. [Google Scholar] [CrossRef]
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef]
- Sun, D.; Wu, H.; Ping, Z.; Zhu, H.; Ai, L. PLAC1 Regulates the Occurrence of Fetal Growth Restriction by Inhibiting the Apoptosis of Trophoblast Cells. Ann. Clin. Lab. Sci. 2021, 51, 182–189. [Google Scholar] [PubMed]
- Filiberto, A.C.; Maccani, M.A.; Koestler, D.; Wilhelm-Benartzi, C.; Avissar-Whiting, M.; Banister, C.E.; Gagne, L.A.; Marsit, C.J. Birthweight is associated with DNA promoter methylation of the glucocorticoid receptor in human placenta. Epigenetics 2011, 6, 566–572. [Google Scholar] [CrossRef]
- Tekola-Ayele, F.; Zeng, X.; Chatterjee, S.; Ouidir, M.; Lesseur, C.; Hao, K.; Chen, J.; Tesfaye, M.; Marsit, C.J.; Workalemahu, T.; et al. Placental multi-omics integration identifies candidate functional genes for birthweight. Nat. Commun. 2022, 13, 2384. [Google Scholar] [CrossRef]
- Teng, L.; Hong, L.J.; Liu, R.Z.; Chen, R.; Li, X.Y.; Yu, M. Cellular Localization and Regulation of Expression of the PLET1 Gene in Porcine Placenta. Int. J. Mol. Sci. 2016, 17, 2048. [Google Scholar] [CrossRef] [PubMed]
- Jeyarajah, M.J.; Jaju Bhattad, G.; Kelly, R.D.; Baines, K.J.; Jaremek, A.; Yang, F.P.; Okae, H.; Arima, T.; Dumeaux, V.; Renaud, S.J. The multifaceted role of GCM1 during trophoblast differentiation in the human placenta. Proc. Natl. Acad. Sci. USA 2022, 119, e2203071119. [Google Scholar] [CrossRef] [PubMed]
- Read, J.E.; Cabrera-Sharp, V.; Kitscha, P.; Cartwright, J.E.; King, P.J.; Fowkes, R.C.; de Mestre, A.M. Glial Cells Missing 1 Regulates Equine Chorionic Gonadotrophin Beta Subunit via Binding to the Proximal Promoter. Front. Endocrinol. 2018, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.L.; Yang, Q.; Zhang, H.; Lin, H.Y.; Zhou, Z.; Lu, X.; Zhu, C.; Xue, L.Q.; Wang, H. Role of placenta-specific protein 1 in trophoblast invasion and migration. Reproduction 2014, 148, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.; Dong, Y. Different expression of lipid metabolism-related genes in Shandong black cattle and Luxi cattle based on transcriptome analysis. Sci. Rep. 2020, 10, 21915. [Google Scholar] [CrossRef]
- Tesfaye, M.; Wu, J.; Biedrzycki, R.J.; Grantz, K.L.; Joseph, P.; Tekola-Ayele, F. Prenatal social support in low-risk pregnancy shapes placental epigenome. BMC Med. 2023, 21, 12. [Google Scholar] [CrossRef]
- Che, L.; Yang, Z.; Xu, M.; Xu, S.; Che, L.; Lin, Y.; Fang, Z.; Feng, B.; Li, J.; Chen, D.; et al. Maternal nutrition modulates fetal development by inducing placental efficiency changes in gilts. BMC Genom. 2017, 18, 213. [Google Scholar] [CrossRef]
- Pei, J.; Zhao, S.; Yin, M.; Wu, F.; Li, J.; Zhang, G.; Wu, X.; Bao, P.; Xiong, L.; Song, W.; et al. Differential proteomics of placentas reveals metabolic disturbance and oxidative damage participate yak spontaneous miscarriage during late pregnancy. BMC Vet. Res. 2022, 18, 248. [Google Scholar] [CrossRef]
- Wawrzykowski, J.; Rapacz-Leonard, A.; Wiacek, D.; Kankofer, M.; Janowski, T. The preliminary studies on protein profile in retained and not retained foetal membranes in heavy draft mares. Reprod. Domest. Anim. 2019, 54, 1543–1551. [Google Scholar] [CrossRef]
- Kwon, S.G.; Hwang, J.H.; Park, D.H.; Kim, T.W.; Kang, D.G.; Kang, K.H.; Kim, I.S.; Park, H.C.; Na, C.S.; Ha, J.; et al. Identification of Differentially Expressed Genes Associated with Litter Size in Berkshire Pig Placenta. PLoS ONE 2016, 11, e0153311. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNAseq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Couture, C.; Brien, M.E.; Boufaied, I.; Duval, C.; Soglio, D.D.; Enninga, E.A.L.; Cox, B.; Girard, S. Proinflammatory changes in the maternal circulation, maternal-fetal interface, and placental transcriptome in preterm birth. Am. J. Obstet. Gynecol. 2023, 228, 332.e1–332.e17. [Google Scholar] [CrossRef]
- Wang, J.; Qian, R.; Wang, Y.; Dong, M.; Liu, X.; Zhou, H.; Ye, Y.; Chen, G.; Chen, D.; Yuan, L.; et al. The mediation effect of placental weight change in the association between prenatal exposure to selenium and birth weight: Evidence from a prospective birth cohort study in China. Environ. Epidemiol. 2021, 5, e139. [Google Scholar] [CrossRef]
- Kramer, A.C.; Steinhauser, C.B.; Gao, H.; Seo, H.; McLendon, B.A.; Burghardt, R.C.; Wu, G.; Bazer, F.W.; Johnson, G.A. Steroids Regulate SLC2A1 and SLC2A3 to Deliver Glucose Into Trophectoderm for Metabolism via Glycolysis. Endocrinology 2020, 16, bqaa098. [Google Scholar] [CrossRef]
- Scheepers, A.; Schmidt, S.; Manolescu, A.; Cheeseman, C.I.; Bell, A.; Zahn, C.; Joost, H.G.; Schürmann, A. Characterization of the human SLC2A11 (GLUT11) gene: Alternative promoter usage, function, expression, and subcellular distribution of three isoforms, and lack of mouse orthologue. Mol. Membr. Biol. 2005, 22, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Elston, R.; Mulligan, C.; Thomas, G.H. Flipping the switch: Dynamic modulation of membrane transporter activity in bacteria. Microbiology 2023, 169, 001412. [Google Scholar] [CrossRef]
- Gyimesi, G.; Hediger, M.A. Transporter-Mediated Drug Delivery. Molecules 2023, 28, 1151. [Google Scholar] [CrossRef]
- Li, T.; Li, X.; Meng, H.; Chen, L.; Meng, F. ACSL1 affects Triglyceride Levels through the PPARγ Pathway. Int. J. Med. Sci. 2020, 17, 720–727. [Google Scholar] [CrossRef]
- Liu, M.; Huang, C.; Dai, R.; Ren, W.; Li, X.; Wu, X.; Ma, X.; Chu, M.; Bao, P.; Guo, X.; et al. Copy Number Variations in the MICALL2 and MOGAT2 Genes Are Associated with Ashidan Yak Growth Traits. Animals 2022, 12, 2779. [Google Scholar] [CrossRef] [PubMed]
- Touzard, E.; Reinaud, P.; Dubois, O.; Guyader-Joly, C.; Humblot, P.; Ponsart, C.; Charpigny, G. Specific expression patterns and cell distribution of ancient and modern PAG in bovine placenta during pregnancy. Reproduction 2013, 146, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, P.M.; Gibson, D.A.; Smith, J.R.; Wilson-Kanamori, J.R.; Kelepouri, O.; Esnal-Zufiaurre, A.; Dobie, R.; Henderson, N.C.; Saunders, P.T.K. Single-cell RNA sequencing redefines the mesenchymal cell landscape of mouse endometrium. FASEB J. 2021, 35, e21285. [Google Scholar] [CrossRef] [PubMed]
- Centenera, M.M.; Scott, J.S.; Machiels, J.; Nassar, Z.D.; Miller, D.C.; Zinonos, I.; Dehairs, J.; Burvenich, I.J.G.; Zadra, G.; Chetta, P.M.; et al. ELOVL5 Is a Critical and Targetable Fatty Acid Elongase in Prostate Cancer. Cancer Res. 2021, 81, 1704–1718. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Abbas Raza, S.H.; Tian, H.; Shi, B.; Luo, Y.; Wang, J.; Liu, X.; Li, S.; Bai, Y.; Hu, J. Effects of overexpression of ACSL1 gene on the synthesis of unsaturated fatty acids in adipocytes of bovine. Arch. Biochem. Biophys. 2020, 695, 108648. [Google Scholar] [CrossRef]
- Joseph, R.; Poschmann, J.; Sukarieh, R.; Too, P.G.; Julien, S.G.; Xu, F.; Teh, A.L.; Holbrook, J.D.; Ng, K.L.; Chong, Y.S.; et al. ACSL1 Is Associated with Fetal Programming of Insulin Sensitivity and Cellular Lipid Content. Mol. Endocrinol. 2015, 29, 909–920. [Google Scholar] [CrossRef]
- Yang, G.; Wang, Y.; Feng, J.; Liu, Y.; Wang, T.; Zhao, M.; Ye, L.; Zhang, X. Aspirin suppresses the abnormal lipid metabolism in liver cancer cells via disrupting an NFκB-ACSL1 signaling. Biochem. Biophys. Res. Commun. 2017, 486, 827–832. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, R.; Zhang, X.; Deng, B.; Mi, C.; Liang, T.; Ling, Y.; Li, H.; Zhang, H. Benzo[a]Pyrene-7, 8-Diol-9, 10-Epoxide Suppresses the Migration and Invasion of Human Extravillous Trophoblast Swan 71 Cells Due to the Inhibited Filopodia Formation and Down-Regulated PI3K/AKT/CDC42/PAK1 Pathway Mediated by the Increased miR-194-3p. Toxicol. Sci. 2018, 166, 25–38. [Google Scholar] [CrossRef]
- Cheng, J.C.; Chang, H.M.; Leung, P.C.K. TGF-β1 Inhibits Human Trophoblast Cell Invasion by Upregulating Connective Tissue Growth Factor Expression. Endocrinology 2017, 158, 3620–3628. [Google Scholar] [CrossRef]
- Suga, N.; Sugimura, M.; Koshiishi, T.; Yorifuji, T.; Makino, S.; Takeda, S. Heparin/heparan sulfate/CD44-v3 enhances cell migration in term placenta-derived immortalized human trophoblast cells. Biol. Reprod. 2012, 86, 1–8. [Google Scholar] [CrossRef]
- Sawai, M.; Uchida, Y.; Ohno, Y.; Miyamoto, M.; Nishioka, C.; Itohara, S.; Sassa, T.; Kihara, A. The 3-hydroxyacyl-CoA dehydratases HACD1 and HACD2 exhibit functional redundancy and are active in a wide range of fatty acid elongation pathways. J. Biol. Chem. 2017, 292, 15538–15551. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.C.; Balas, L.; Bazan, N.G.; Brenna, J.T.; Chiang, N.; da Costa Souza, F.; Dalli, J.; Durand, T.; Galano, J.M.; Lein, P.J.; et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog. Lipid Res. 2022, 86, 101165. [Google Scholar] [CrossRef] [PubMed]
- Duttaroy, A.K.; Basak, S. Maternal Fatty Acid Metabolism in Pregnancy and Its Consequences in the Feto-Placental Development. Front. Physiol. 2022, 12, 787848. [Google Scholar] [CrossRef]
- Deng, M.; Du, S.; Hou, H.; Xiao, J. Structural insights into the high-affinity IgE receptor FcεRI complex. Nature 2024, 1–8. [Google Scholar] [CrossRef]
- Vitte, J.; Vibhushan, S.; Bratti, M.; Montero-Hernandez, J.E.; Blank, U. Allergy, Anaphylaxis, and Nonallergic Hypersensitivity: IgE, Mast Cells, and Beyond. Med. Princ. Pract. 2022, 31, 501–515. [Google Scholar] [CrossRef]
- Nguyen, S.M.T.; Rupprecht, C.P.; Haque, A.; Pattanaik, D.; Yusin, J.; Krishnaswamy, G. Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond. Int. J. Mol. Sci. 2021, 22, 7785. [Google Scholar] [CrossRef]
- Porcherie, A.; Mathieu, C.; Peronet, R.; Schneider, E.; Claver, J.; Commere, P.H.; Kiefer-Biasizzo, H.; Karasuyama, H.; Milon, G.; Dy, M.; et al. Critical role of the neutrophil-associated high-affinity receptor for IgE in the pathogenesis of experimental cerebral malaria. J. Exp. Med. 2011, 208, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, M.; Mizutani, S.; Matsumoto, K.; Kato, Y.; Masuo, Y.; Harumasa, A.; Iyoshi, S.; Tano, S.; Mizutani, H.; Kotani, T.; et al. The balance between fetal oxytocin and placental leucine aminopeptidase (P-LAP) controls human uterine contraction around labor onset. Eur. J. Obstet. Gynecol. Reprod. Biol. X. 2023, 19, 100210. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, J.; Chen, Y.; Guo, J.; Li, Q.; Dinnyes, A.; Sun, Q.; Liu, X.; He, G.; Zhu, B.; et al. Placenta-specific CYP11A1 overexpression lead to autism-like symptom in offspring with altered steroid hormone biosynthesis in the placenta-brain axis and rescued by vitamin D intervention. Brain Behav. Immun. 2024, 121, 13–25. [Google Scholar] [CrossRef]
- Pan, T.; He, G.; Chen, M.; Bao, C.; Chen, Y.; Liu, G.; Zhou, M.; Li, S.; Xu, W.; Liu, X. Abnormal CYP11A1 gene expression induces excessive autophagy, contributing to the pathogenesis of preeclampsia. Oncotarget 2017, 8, 89824–89836. [Google Scholar] [CrossRef]
- Yoshihara, M.; Mizutani, S.; Matsumoto, K.; Kato, Y.; Masuo, Y.; Tano, S.; Mizutani, H.; Kotani, T.; Mizutani, E.; Shibata, K.; et al. Crosstalk between foetal vasoactive peptide hormones and placental aminopeptidases regulates placental blood flow: Its significance in preeclampsia. Placenta 2022, 121, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Peterson, H.F.; Eskild, A.; Sommerfelt, S.; Hillestad, V. Placental size at gestational week 36: Comparisons between ongoing pregnancies and deliveries. Acta Obstet. Gynecol. Scand. 2024, 103, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Huang, P.; Miao, H.; Yu, H.; Wu, D.; Chen, F.; Lin, Y.; Lin, Y.; Li, W.; Hua, J. The new landscape of differentially expression proteins in placenta tissues of gestational diabetes based on iTRAQ proteomics. Placenta 2023, 131, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.M.; Wadsack, C.; Desoye, G. Placental fatty acid transfer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Herrera, E.; Amusquivar, E.; López-Soldado, I.; Ortega, H. Maternal lipid metabolism and placental lipid transfer. Horm. Res. 2006, 65 (Suppl. 3), 59–64. [Google Scholar] [CrossRef] [PubMed]
- Yañez, M.J.; Leiva, A. Human Placental Intracellular Cholesterol Transport: A Focus on Lysosomal and Mitochondrial Dysfunction and Oxidative Stress. Antioxidants 2022, 11, 500. [Google Scholar] [CrossRef]
- Mistry, H.D.; Kurlak, L.O.; Mansour, Y.T.; Zurkinden, L.; Mohaupt, M.G.; Escher, G. Increased maternal and fetal cholesterol efflux capacity and placental CYP27A1 expression in preeclampsia. J. Lipid Res. 2017, 58, 1186–1195. [Google Scholar] [CrossRef]
- Fu, Y.T.; Zhang, J.; Liu, W.B.; Zhang, Y.F.; Zhang, S.; Tan, L.L.; Lin, Q.; Ou-Yang, K.W.; Xiong, Y.W.; Chang, W.; et al. Gestational cadmium exposure disrupts fetal liver development via repressing estrogen biosynthesis in placental trophoblasts. Food Chem. Toxicol. 2023, 176, 113807. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Liu, R.; Chen, Z.; Zhang, R.; Mei, Y.; Miao, X.; Bai, X.; Dong, Y. Combining Transcriptomics and Proteomics to Screen Candidate Genes Related to Bovine Birth Weight. Animals 2024, 14, 2751. https://doi.org/10.3390/ani14182751
Wang X, Liu R, Chen Z, Zhang R, Mei Y, Miao X, Bai X, Dong Y. Combining Transcriptomics and Proteomics to Screen Candidate Genes Related to Bovine Birth Weight. Animals. 2024; 14(18):2751. https://doi.org/10.3390/ani14182751
Chicago/Turabian StyleWang, Xiuyuan, Ruili Liu, Zhenpeng Chen, Renzheng Zhang, Yanfang Mei, Xiuping Miao, Xuejin Bai, and Yajuan Dong. 2024. "Combining Transcriptomics and Proteomics to Screen Candidate Genes Related to Bovine Birth Weight" Animals 14, no. 18: 2751. https://doi.org/10.3390/ani14182751
APA StyleWang, X., Liu, R., Chen, Z., Zhang, R., Mei, Y., Miao, X., Bai, X., & Dong, Y. (2024). Combining Transcriptomics and Proteomics to Screen Candidate Genes Related to Bovine Birth Weight. Animals, 14(18), 2751. https://doi.org/10.3390/ani14182751