

Genetic Structure and Population History of the Zaisan Toad-Headed Agama (Phrynocephalus melanurus) Inferred from Mitochondrial DNA
 , , , ,
, , , , 
Abstract
Simple Summary
Abstract
1. Introduction

2. Materials and Methods
2.1. Population Sampling
2.2. DNA Isolation, PCR Amplification, and Sequencing
2.3. Phylogenetic Reconstruction
2.4. Divergence Times Estimation
2.5. Genetic Diversity and Population Genetic Structure Analyses
2.6. Inference of Demographic History
2.7. Phylogeographic Diffusion in Continuous Space
2.8. Past, Present, and Future Distribution Models
3. Results
3.1. Sequence Characteristics
3.2. Phylogenetic Relationships
3.3. Timeframe of the Diversification
3.4. Population Genetic Structure
3.5. Historical Demography
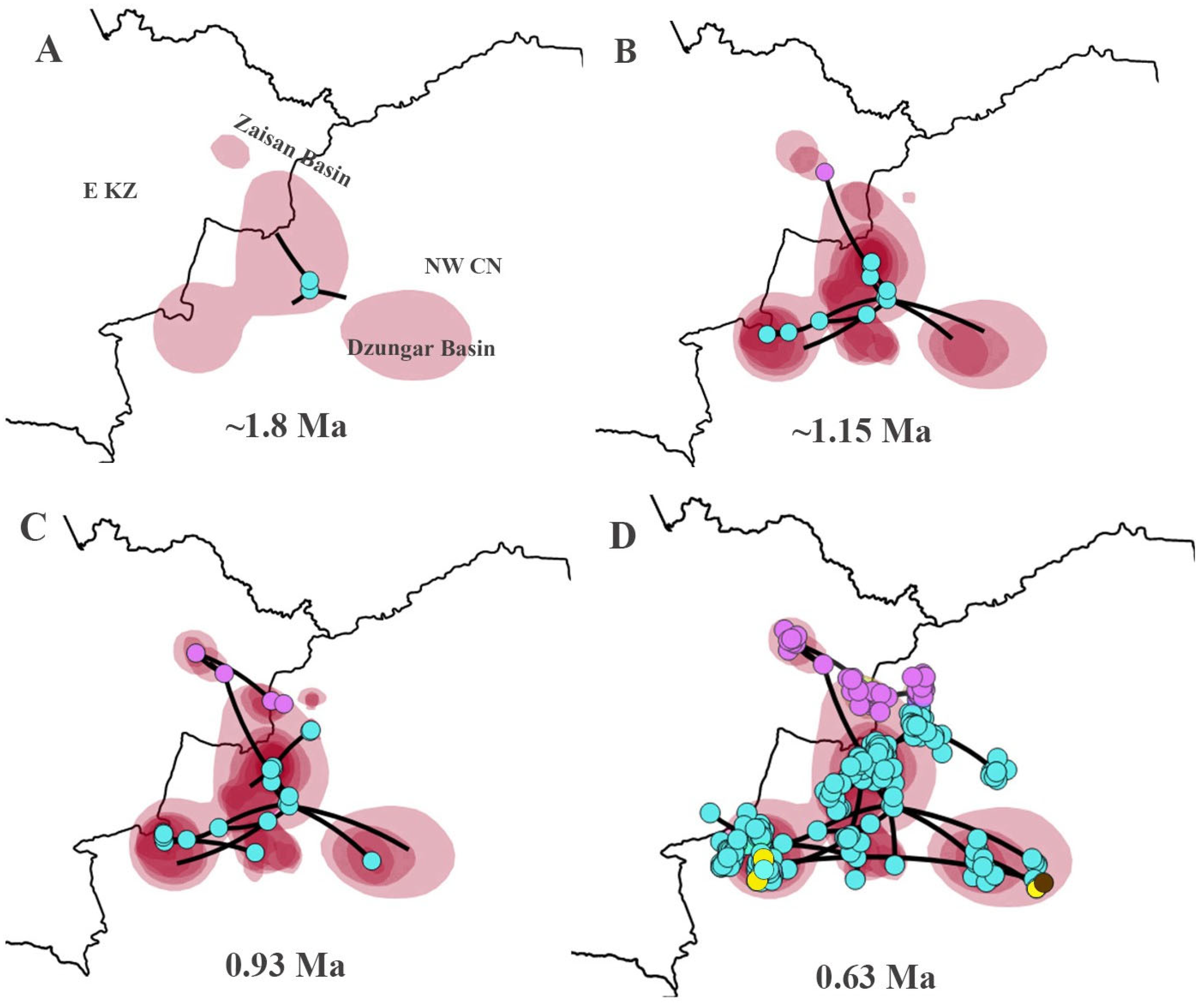
3.6. The Spatiotemporal Diffusion for P. melanurus
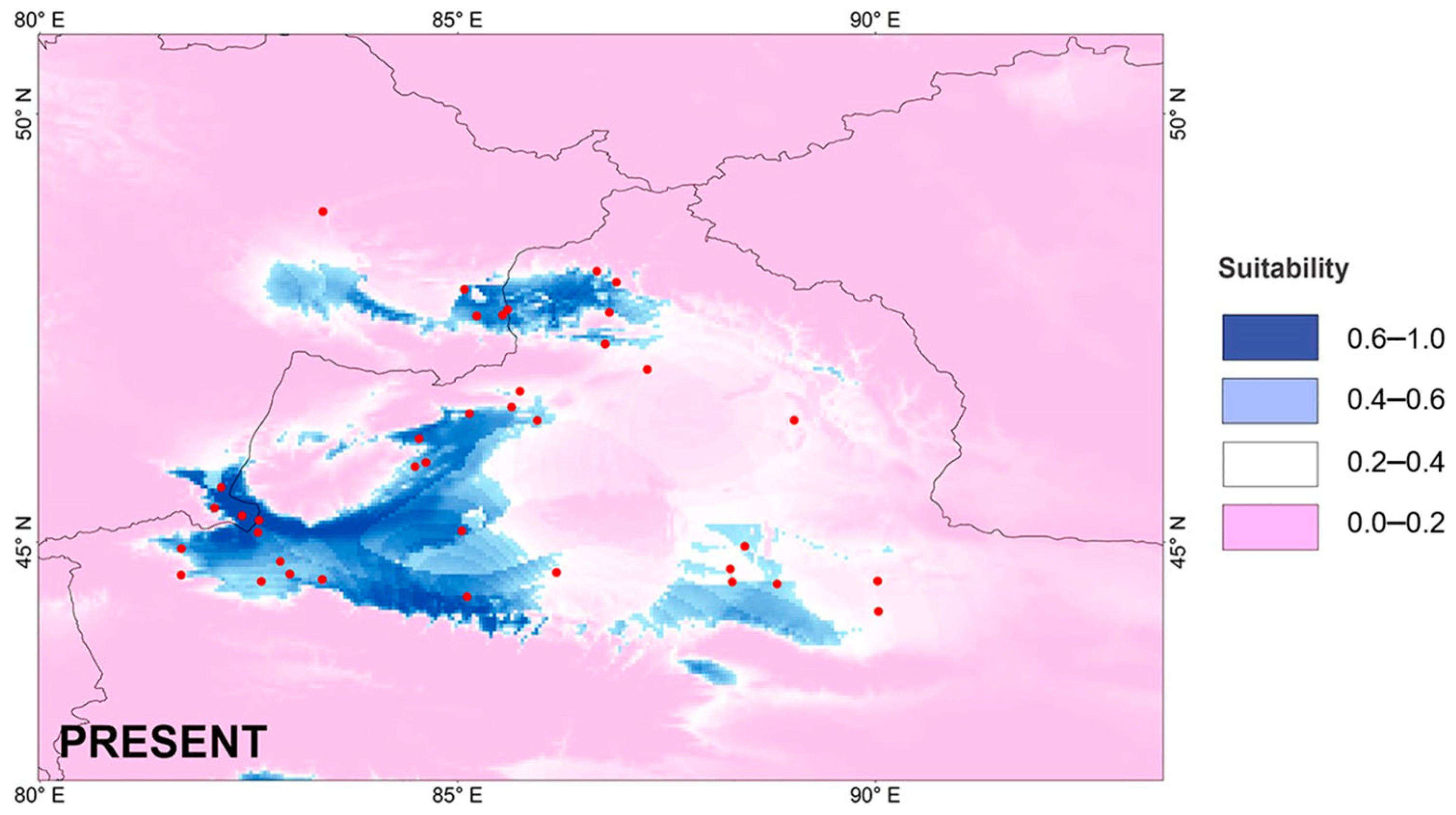
3.7. Potential Species Distribution Modeling
4. Discussion
4.1. Phylogeographic Pattern and Diversification History
4.2. Origin and Colonization
4.3. Lineage-Specific Response to Quaternary Climatic Oscillations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Byrne, M.; Yeates, D.K.; Joseph, L.; Kearney, M.; Bowler, J.; Williams, M.A.J.; Coper, S.; Donnellan, S.C.; Keogh, J.S.; Leys, R.; et al. Birth of a biome: Insights into the assembly and maintenance of the Australian arid zone biota. Mol. Ecol. 2008, 17, 4398–4417. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.P.; Pendergrass, A.G.; Rapp, A.D.; Wodzicki, K.R. Response of the intertropical convergence zone to climate change: Location, width, and strength. Curr. Clim. Chang. Rep. 2018, 4, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Melville, J.; Hale, J.; Mantziou, G.; Ananjeva, N.B.; Milto, K.; Clemann, N. Historical biogeography, phylogenetic relationships and intraspecific diversity of agamid lizards in the Central Asian deserts of Kazakhstan and Uzbekistan. Mol. Phylogenet. Evol. 2009, 53, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.M.; Ji, Y.J.; Liu, L.; Wang, L.; Zhang, D.X. Impact of climate changes from Middle Miocene onwards on evolutionary diversification in Eurasia: Insights from the mesobuthid scorpions. Mol. Ecol. 2013, 22, 1700–1716. [Google Scholar] [CrossRef] [PubMed]
- Kearns, A.; Joseph, L.; Toon, A.; Cook, L.G. Australia’s arid-adapted butcherbirds experienced range expansions during Pleistocene glacial maxima. Nat. Commun. 2014, 5, 3994. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.D.; Zhang, L.J.; Lin, L.; Yu, S.; Chen, Z.; Wang, W. Insights into the historical assembly of global dryland floras: The diversification of Zygophyllaceae. BMC Evol. Biol. 2018, 18, 166. [Google Scholar] [CrossRef] [PubMed]
- Riddle, B.R.; Hafner, D.J. A step-wise approach to integrating phylogeographic and phylogenetic biogeographic perspectives on the history of a core North American warm deserts biota. J. Arid Environ. 2006, 66, 435–461. [Google Scholar] [CrossRef]
- Liu, J.; Guo, X.; Chen, D.; Li, J.; Yue, B.; Zeng, X. Diversification and historical demography of the rapid racerunner (Eremias velox) in relation to geological history and Pleistocene climatic oscillations in arid Central Asia. Mol. Phylogenet. Evol. 2019, 130, 244–258. [Google Scholar] [CrossRef]
- Maestre, F.T.; Benito, B.M.; Berdugo, M.; Concostrina-Zubiri, L.; Delgado-Baquerizo, M.; Eldridge, D.J.; Guirado, E.; Gross, N.; Kéfi, S.; Le Bagousse-Pinguet, Y.; et al. Biogeography of global drylands. New Phytol. 2021, 231, 540–558. [Google Scholar] [CrossRef]
- Shi, C.M.; Zhang, X.S.; Liu, L.; Ji, Y.J.; Zhang, D.X. Phylogeography of the desert scorpion illuminates a route out of Central Asia. Curr. Zool. 2023, 69, 442–455. [Google Scholar] [CrossRef]
- Wu, N.; Wang, S.; Dujsebayeva, T.; Chen, D.; Ali, A.; Guo, X. Geography and past climate changes have shaped the evolution of a widespread lizard in arid Central Asia. Mol. Phylogenet. Evol. 2023, 184, 107781. [Google Scholar] [CrossRef] [PubMed]
- An, Z.S.; Kutzbach, J.E.; Prell, W.L.; Porter, S.C. Evolution of Asian monsoons and phased uplift of the Himalaya-Tibetan plateau since Late Miocene times. Nature 2001, 411, 62–66. [Google Scholar] [CrossRef]
- Guo, Z.T.; Ruddiman, W.F.; Hao, Q.Z.; Wu, H.B.; Qiao, Y.S.; Zhu, R.X.; Peng, S.Z.; Wei, J.J.; Yuan, B.Y.; Liu, T.S. Onset of Asian desertification by 22 Myr ago inferred from loess deposits in China. Nature 2002, 416, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ye, J.; Wu, W.; Ni, X.; Bi, S.; Zhang, Z.; Liu, W.; Meng, J. Late Oligocene-Miocene mid-latitude aridification and wind patterns in the Asian interior. Geology 2010, 38, 515–518. [Google Scholar] [CrossRef]
- Lu, H.; Wang, X.; Wang, X.; Chang, X.; Zhang, H.; Xu, Z.; Zhang, W.; Wei, H.; Zhang, X.; Yi, S.; et al. Formation and evolution of Gobi Desert in central and eastern Asia. Earth Sci. Rev. 2019, 194, 251–263. [Google Scholar] [CrossRef]
- Fang, X.; Lu, L.; Yang, S.; Li, J.; An, Z.; Jiang, P.; Chen, X. Loess in Kunlun Mountains and its implications on desert development and Tibetan Plateau uplift in west China. Sci. China Ser. D 2002, 45, 289–299. [Google Scholar] [CrossRef]
- Miao, Y.; Herrmann, M.; Wu, F.; Yan, X.; Yang, S. What controlled Mid–Late Miocene long-term aridification in Central Asia?—Global cooling or Tibetan Plateau uplift: A review. Earth-Sci. Rev. 2012, 112, 155–172. [Google Scholar] [CrossRef]
- Li, Z.H.; Chen, J.; Zhao, G.F.; Guo, Y.P.; Kou, Y.X.; Ma, Y.Z.; Wang, G.; Ma, X.F. Response of a desert shrub to past geological and climatic change: A phylogeographic study of Reaumuria soongarica (Tamaricaceae) in western China. J. Syst. Evol. 2012, 50, 351–361. [Google Scholar] [CrossRef]
- Meng, H.H.; Zhang, M.L. Diversification of plant species in arid northwest China: Species-level phylogeographic history of Lagochilus Bunge ex Bentham (Lamiaceae). Mol. Phylogenet. Evol. 2013, 68, 389–409. [Google Scholar] [CrossRef]
- Su, S.H.; Zhang, M.L. A range wide geographic pattern of genetic diversity and population structure of Hexinia polydichotoma (Asteraceae) in Tarim Basin and adjacent areas. Biochem. Syst. Ecol. 2014, 56, 49–59. [Google Scholar] [CrossRef]
- Nanova, O.G.; Lebedev, V.S.; Matrosova, V.A.; Adiya, Y.; Undrakhbayar, E.; Surov, A.V.; Shenbrot, G.I. Phylogeography, phylogeny, and taxonomical revision of the Midday jird (Meriones meridianus) species complex from Dzungaria. J. Zool. Syst. Evol. Res. 2020, 58, 1335–1358. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, Y.; Li, W.; Fu, A.; Ma, X.; Gui, D.; Chen, Y. Distribution pattern of plant species diversity in the mountainous region of Ili River Valley, Xinjiang. Environ. Monit. Assess. 2011, 177, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.H.; Gao, X.Y.; Huang, J.F.; Zhang, M.L. Plant phylogeography in arid Northwest China: Retrospectives and perspectives. J. Syst. Evol. 2015, 53, 33–46. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, H.-X. Population genetic structure and biodiversity conservation of a relict and medicinal subshrub Capparis spinosa in arid Central Asia. Diversity 2022, 14, 146. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, G.; Feng, Y.; Zhang, D.; Shi, C. Genetic analysis and ecological niche modeling delimit species boundary of the Przewalski’s scorpion (Scorpions: Buthidae) in arid Asian inland. Zool. Syst. 2020, 45, 81–96. [Google Scholar] [CrossRef]
- Camargo, A.; Werneck, F.P.; Morando, M.; Sites, J.W., Jr.; Avila, L.J. Quaternary range and demographic expansion of Liolaemus darwinii (Squamata: Liolaemidae) in the Monte Desert of Central Argentina using Bayesian phylogeography and ecological niche modelling. Mol. Ecol. 2013, 22, 4038–4054. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.N.; Barley, A.J.; Poe, S.; Thomson, R.C.; Nieto-Montes de Oca, A.; Wang, I.J. Phylogeography of a widespread lizard complex reflects patterns of both geographic and ecological isolation. Mol. Ecol. 2019, 28, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wen, L. Impacts of Quaternary glaciation, geological history and geography on animal species history in continental East Asia: A phylogeographic review. Mol. Ecol. 2023, 32, 4497–4514. [Google Scholar] [CrossRef]
- Golubev, M.L. Three controversial questions of systematics and nomenclature in Phrynocephalus of USSR fauna (Phrynocephalus, Agamidae). In Questions of Herpetology: Abstracts of the 7th Conference; Naukova Dumka: Kiev, Ukraine, 1989; pp. 44–45. (In Russian) [Google Scholar]
- Golubev, M.L. Variegated toad-head agama Phrynocephalus versicolor (Reptilia, Agamidae) in Dzungar gates (East Kazakhstan) with notes on the species systematics. Herald Zool. 1992, 2, 31–238. (In Russian) [Google Scholar]
- Dunayev, E.A. Systematic condition, ecological and behavioral peculiarities of Zaisan toad-headed agama Phrynocephalus melanurus Eichwald, 1831 (Reptilia: Agamidae). Bull. Moscow Soc. Nat. Ser. Bio. 1989, 94, 41–53. (In Russian) [Google Scholar]
- Dunayev, E.A. Systematics and Paleogeography: Conceptual Synthesis Using an Example of Phrynocephalus (superspecies guttatus) (Reptilia: Agamidae). In Evolution and Systematics: Lamark and Darwin in Modern Research: Proceedings of the Zoological Museum of Moscow State University; KMK Scientific Press: Moscow, Russia, 2009; Volume 50, pp. 275–298. (In Russian) [Google Scholar]
- Barabanov, A.V.; Ananjeva, N.B. Catalogue of the available scientific species-group names for lizards of the genus Phrynocephalus Kaup, 1825 (Reptilia, Sauria, Agamidae). Zootaxa 2007, 1399, 1–56. [Google Scholar] [CrossRef]
- Dunayev, E.A.; Solovyova, E.N.; Poyarkov, N.A. Taxonomy, phylogeny and distribution of Phrynocephalus (superspecies guttatus) (Reptilia: Agamidae). Curr. Stud. Herpetol. 2020, 20, 16–34. (In Russian) [Google Scholar] [CrossRef]
- Dunayev, E.A. On the possible use of the ethological features in the taxonomy and phylogeny of toad agamas. Russ. J. Herpetol. 1996, 3, 32–38. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Nahbolz, B.; Glemin, S.; Hurst, G.D. Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 2009, 18, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Solovyeva, E.N.; Poyarkov, N.A.; Dunayev, E.A.; Nazarov, R.A.; Lebedev, V.S.; Bannikova, A.A. Phylogenetic relationships and subgeneric taxonomy of toad headed agamas Phrynocephalus (Reptilia, Squamata, Agamidae) as determined by mitochondrial DNA sequencing. Dokl. Biol. Sci. 2014, 455, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Aljanabi, S.M.; Martinez, I. Universal and rapid salt-extraction of high-quality genomic DNA for PCR-based techniques. Nucl. Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef]
- Solovyova, E.N.; Lebedev, V.S.; Dunayev, E.A.; Nazarov, R.A.; Bannikova, A.A.; Che, J.; Murphy, R.W.; Poyarkov, N.A. Cenozoic aridization in central Eurasia shaped diversification of toad-headed agamas (Phrynocephalus; Agamidae, Reptilia). PeerJ 2018, 6, 4543. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Rannala, B. Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. Syst. Biol. 2004, 53, 904–913. [Google Scholar] [CrossRef]
- FigTree v1.4.4. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 21 April 2021).
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987; 512p. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. PopART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef] [PubMed]
- Godbout, J.; Jaramillo-Correa, J.P.; Beaulieu, J. A mitochondrial DNA minisatellite reveals the postglacial history of jack pine (Pinus banksiana), a broad range North American conifer. Mol. Ecol. 2005, 141, 3497–3512. [Google Scholar] [CrossRef]
- Magri, D.; Vendramin, G.G.; Comps, B.; Dupanloup, I.; Geburek, T.; Gömöry, D.; Latałowa, M.; Litt, T.; Paule, L.; Roure, J.M.; et al. A new scenario for the Quaternary history of European beech populations: Palaeobotanical evidence and genetic consequences. New Phytol. 2006, 171, 199–221. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. Vegan, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Ramos-Onsins, S.E.; Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 2002, 19, 2092–2100. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J. Bayesian Evolutionary Analysis of Viruses: A Practical Introduction to BEAST; The Pasteur Institute: Paris, France, 2009; 34p. [Google Scholar]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- GBIF.org (24 April 2023) GBIF Occurrence Download. 24 April. [CrossRef]
- Brown, J.L. SDMtoolbox: A python-based GIS toolkit for landscape genetic, biogeographic and species distribution model analyses. Methods Ecol. Evol. 2014, 5, 694–700. [Google Scholar] [CrossRef]
- Karger, D.N.; Conrad, O.; Böhner, J.; Kawohl, T.; Kreft, H.; Soria-Auza, R.W.; Zimmermann, N.E.; Kessler, M. Data Descriptor: Climatologies at high resolution for the earth’s land surface areas. Sci. Data 2017, 4, 170122. [Google Scholar] [CrossRef]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high-resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Synes, N.W.; Osborne, P.E. Choice of predictor variables as a source of uncertainty in continental-scale species distribution modelling under climate change. Glob. Ecol. Biogeogr. 2011, 20, 904–914. [Google Scholar] [CrossRef]
- Shiogama, H.; Tatebe, H.; Hayashi, M.; Abe, M.; Arai, M.; Koyama, H.; Imada, Y.; Kosaka, Y.; Ogura, T.; Watanabe, M. MIROC6 Large Ensemble (MIROC6-LE): Experimental design and initial analyses. Earth Syst. Dynam. 2023, 14, 1107–1124. [Google Scholar] [CrossRef]
- Eyring, V.; Bony, S.; Meehl, G.A.; Senior, C.A.; Stevens, B.; Stouffer, R.J.; Taylor, K.E. Overview of the Coupled Model Intercomparison Project Phase 6 (CMIP6) experimental design and organization. Geosci. Model Dev. 2016, 9, 1937–1958. [Google Scholar] [CrossRef]
- Kriegler, E.; O’Neill, B.C.; Hallegatte, S.; Kram, T.; Lempert, R.J.; Moss, R.H.; Wilbanks, T. The need for and use of socio-economic scenarios for climate change analysis: A new approach based on shared socio-economic pathways. Glob. Environ. Chang. 2012, 22, 807–822. [Google Scholar] [CrossRef]
- Phillips, S.B.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Muscarella, R.; Galante, P.J.; Soley-Guardia, M.; Boria, R.A.; Kass, J.M.; Uriarte, M.; Anderson, R.P. ENMeval: An R package for conducting spatially independent evaluations and estimating optimal model complexity for Maxent ecological niche models. Methods Ecol. Evol. 2014, 5, 1198–1205. [Google Scholar] [CrossRef]
- Phillips, S.J.; Dudik, M. Modeling of species distributions with Maxent: New extensions and a comprehensive evaluation. Ecography 2008, 31, 161–175. [Google Scholar] [CrossRef]
- Elith, J.; Kearney, M.; Phillips, S. The art of modelling range-shifting species. Methods Ecol. Evol. 2010, 1, 330–342. [Google Scholar] [CrossRef]
- Macey, J.R.; Schulte, J.A.; Ananjeva, N.B.; Van Dyke, E.T.; Wang, Y.; Orlov, N.; Shafiei, S.; Robinson, M.D.; Dujsebayeva, T.; Freund, G.S.; et al. A molecular phylogenetic hypothesis for the Asian agamid lizard genus Phrynocephalus reveals discrete biogeographic clades implicated by plate tectonics. Zootaxa 2018, 4467, 1–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fu, J. Cladogenesis and vicariance patterns in the toad-headed lizard Phrynocephalus versicolor species complex. Copeia 2004, 2004, 199–206. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, M.-L. Phylogeography of the arid shrub Atrafaxis frutescens (Polygonacea) on Northwest China: Evidence from cpDNA sequences. J. Hered. 2015, 106, 184–195. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.-L.; Yin, L.-K. Phylogeographic structure of a Tethyan relict Capparis spinosa (Capparaceae) traces Pleistocene geologic and climatic changes in the Western Himalayas, Tianshan Mountains, and adjacent desert regions. BioMed Res. Int. 2016, 2016, 5792708. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Wang, Z.X.; Jin, X.C.; Li, T.; Li, Y. Evolution, chronology and depositional effect of uplifting in the eastern sector of the Tianshan Mountains. Acta Geol. Sin. 2004, 78, 319–331, (In Chinese with English Abstract). [Google Scholar] [CrossRef]
- Sun, J.M.; Zhu, R.X. Cenozoic deposits in the Northern Tianshan mountains and its implications for neotectonics and environmental changes. Quatern. Sci. 2006, 1, 14–19, (In Chinese with English Abstract). [Google Scholar] [CrossRef]
- Shi, Y.-F.; Cui, Z.-J.; Su, Z. The Quaternary Glaciations and Environmental Variations in China; Hebei Science and Technology Press: Shijiazhuang, China, 2006. (In Chinese) [Google Scholar]
- Nicolas, V.; Mboumba, J.F.; Verheyen, E.; Denys, C.; Lecompte, E.; Olayemi, A.; Missoup, A.D.; Katuala, P.; Colyn, M. Phylogeographic structure and regional history of Lemniscomys striatus (Rodentia: Muridae) in tropical Africa. J. Biogeogr. 2008, 35, 2074–2089. [Google Scholar] [CrossRef]
- Brouat, C.; Tatard, C.; Bâ, K.; Cosson, J.F.; Dobigny, G.; Fichet-Calvet, E.; Granjon, L.; Lecompte, E.; Loiseau, A.; Mouline, K.; et al. Phylogeography of the Guinea multimammate mouse (Mastomys erythroleucus): A case study for Sahelian species in West Africa. J. Biogeogr. 2009, 36, 2237–2250. [Google Scholar] [CrossRef]
- Svarichevskaya, Z.A. Geomorphology of Kazakhstan and Middle Asia; LSU: Leningrad, Russia, 1965; 296p. (In Russian) [Google Scholar]
- Shyukin, I.S. Geomorphology of Central Asia; MSU: Moscow, Russia, 1983; 432p. (In Russian) [Google Scholar]
- Galbreath, K.E.; Hafner, D.J.; Zamudio, K.R. When cold is better: Climate-driven elevation shifts yield complex patterns of diversification and demography in an alpine specialist (American pika, Ochotona princeps). Evolution 2009, 63, 2848–2863. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yi, S.; Xu, Z.; Zhou, Y.; Zeng, L.; Zhu, F.; Feng, H.; Dong, L.; Zhuo, H.; Yu, K.; et al. Chinese deserts and sand fields in Last Glacial Maximum and Holocene Optimum. Chin. Sci. Bull. 2013, 58, 2775–2783. [Google Scholar] [CrossRef]
- Taberlet, P.; Fumagalli, L.; Wust–Saucy, A.G.; Cosson, J.F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef]
- Petit, R.J.; Aguinagalde, I.; de Beaulieu, J.L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M.; et al. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef]
- Provan, J.; Bennett, K.D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 2008, 23, 564–571. [Google Scholar] [CrossRef]
- Stewart, J.R.; Lister, A.M.; Barnes, I.; Dalén, L. Refugia revisited: Individualistic responses of species in space and time. Proc. Biol. Sci. 2009, 277, 661–671. [Google Scholar] [CrossRef]
- McCain, C.M.; King, S.R.B. Body size and activity times mediate mammalian responses to climate change. Glob. Chang. Biol. 2014, 20, 1760–1769. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site Number | Locality | N | X | Y | Genetic Diversity | ||
|---|---|---|---|---|---|---|---|
| Haplotypes | Hd ± SD | ± SD | |||||
| 1 | Ulungur, CN | 5 | 87.26 | 47.01 | H6, H12–13 | 0.700 ± 0.218 | 0.00254 ± 0.00079 |
| 2 | Karamay 1, CN | 9 | 85.95 | 46.41 | H6, H13, H32–35 | 0.889 ± 0.091 | 0.00300 ± 0.00073 |
| 3 | Karamay 2, CN | 4 | 85.05 | 45.13 | H6–7 | 0.500 ± 0.07031 | 0.00873 ± 0.00463 |
| 4 | Hoboksar 1, CN | 11 | 85.74 | 46.76 | H13, H42 | 0.509 ± 0.101 | 0.00485 ± 0.00096 |
| 5 | Hoboksar 2, CN | 6 | 85.64 | 46.57 | H6, H36–37 | 0.733 ± 0.155 | 0.00180 ± 0.00039 |
| 6 | Hoboksar 3, CN | 10 | 85.14 | 46.50 | H6, H38–40, H48 | 0.844 ± 0.080 | 0.00317 ± 0.00052 |
| 7 | Emin, CN | 1 | 84.53 | 46.20 | H7 | – | – |
| 8 | Toli 1, CN | 3 | 84.49 | 45.88 | H4–5 | 0.667 ± 0.314 | 0.00529 ± 0.00249 |
| 9 | Toli 2, CN | 2 | 84.62 | 45.93 | H4 | 0.000 ± 0.000 | 0.00000 ± 0.00000 |
| 10 | Fuyun, CN | 6 | 89.02 | 46.42 | H6 | 0.000 ± 0.000 | 0.00000 ± 0.00000 |
| 11 | Jeminay, CN | 13 | 86.76 | 47.31 | H6, H39. H43–47, H49 | 0.808 ± 0.113 | 0.00265 ± 0.00061 |
| 12 | Shihezi, CN | 1 | 86.18 | 44.64 | H1 | – | – |
| 13 | Fukang 1, CN | 4 | 88.43 | 44.95 | H16–18 | 0.833 ± 0.222 | 0.00185 ± 0.00058 |
| 14 | Fukang 2, CN | 3 | 88.28 | 44.53 | H17, H21 | 0.667 ± 0.314 | 0.00317 ± 0.00150 |
| 15 | Fukang 3, CN | 2 | 88.26 | 44.68 | H21 | – | – |
| 16 | Jimsar, CN | 4 | 88.82 | 44.51 | H16, H22–23 | 0.833 ± 0.222 | 0.00265 ± 0.00071 |
| 17 | Ebinur 1, CN | 2 | 82.61 | 45.11 | H28, H31 | 1.000 ± 0.500 | 0.00476 ± 0.00238 |
| 18 | Ebinur 2, CN | 1 | 82.87 | 44.77 | H25 | – | – |
| 19 | Bortala 1, CN | 7 | 81.69 | 44.61 | H24, H26 | 0.286 ± 0.196 | 0.00045 ± 0.00031 |
| 20 | Bortala 2, CN | 1 | 82.60 | 45.19 | H3 | – | – |
| 21 | Kuytun, CN | 1 | 85.11 | 44.36 | H14 | – | – |
| 22 | Bole, CN | 11 | 81.69 | 44.92 | H24, H27–30 | 0.709 ± 0.099 | 0.00404 ± 0.00056 |
| 23 | Qitai 1, CN | 1 | 90.03 | 44.19 | H16 | – | – |
| 24 | Qitai 2, CN | 4 | 90.02 | 44.54 | H2, H19–20 | 0.833 ± 0.222 | 0.01138 ± 0.00554 |
| 25 | Alashankou, CN | 9 | 82.62 | 45.25 | H3, H28, H31 | 0.556 ± 0.165 | 0.00132 ± 0.00055 |
| 26 | Jinghe 1, CN | 11 | 82.65 | 44.54 | H2, H15 | 0.182 ± 0.144 | 0.00029 ± 0.00023 |
| 27 | Jinghe 2, CN | 4 | 82.58 | 44.54 | H2, H3 | 0.500 ± 0.265 | 0.00635 ± 0.00337 |
| 28 | Jinghe 3, CN | 1 | 82.99 | 44.62 | H3 | – | – |
| 29 | Jinghe 4, CN | 1 | 83.37 | 44.56 | H3 | – | – |
| 30 | Bolade, CN | 1 | 86.66 | 48.16 | H11 | – | – |
| 31 | Buerjin 1, CN | 5 | 86.90 | 48.03 | H10 | 0.000 ± 0.000 | 0.00000 ± 0.00000 |
| 32 | Buerjin 2, CN | 9 | 86.81 | 47.69 | H9 | 0.000 ± 0.000 | 0.00000 ± 0.00000 |
| 33 | Zaisan, KZ | 4 | 85.59 | 47.71 | H8, H9 | 0.667 ± 0.204 | 0.00106 ± 0.00032 |
| 34 | Kurchum, KZ | 5 | 85.08 | 47.94 | H9, H50 | 0.400 ± 0.237 | 0.00127 ± 0.00075 |
| 35 | Kokpekty 1, KZ | 1 | 83.38 | 48.85 | H50 | – | – |
| 36 | Kokpekty 2, KZ | 2 | 83.42 | 48.80 | H50 | 0.000 ± 0.000 | 0.00000 ± 0.00000 |
| Clade/Subclade | N | Nh | S | m | K | Hd ± SD | π ± SD |
|---|---|---|---|---|---|---|---|
| I | 27 | 6 | 7 | 7 | 1.13390 | 0.661 ± 0.086 | 0.00180 ± 0.00038 |
| II | 138 | 44 | 57 | 60 | 5.95261 | 0.942 ± 0.010 | 0.00945 ± 0.00048 |
| IIa | 3 | 2 | 1 | 1 | 0.66667 | 0.667 ± 0.314 | 0.00106 ± 0.00050 |
| IIb1 | 15 | 2 | 1 | 1 | 0.13333 | 0.133 ± 0.112 | 0.00021 ± 0.00018 |
| IIb2 | 14 | 6 | 6 | 6 | 1.89011 | 0.857 ± 0.056 | 0.00300 ± 0.00029 |
| IIb3 | 70 | 24 | 29 | 31 | 2.46253 | 0.863 ± 0.034 | 0.00391 ± 0.00040 |
| All | 165 | 50 | 64 | 69 | 9.08914 | 0.950 ± 0.00006 | 0.01443 ± 0.00075 |
| Clade/Subclade | I | IIa | IIb1 | IIb2 | IIb3 |
|---|---|---|---|---|---|
| I | 0.00550 | 0.00626 | 0.00570 | 0.00585 | |
| IIa | 0.02534 | 0.00562 | 0.00519 | 0.00517 | |
| IIb1 | 0.03017 | 0.02180 | 0.00418 | 0.00432 | |
| IIb2 | 0.02699 | 0.02033 | 0.01463 | 0.00264 | |
| IIb3 | 0.02874 | 0.02165 | 0.01539 | 0.01095 |
| Clade/Subclade | Tajima’s D | Fu’s Fs | R2 | Rg | SSD |
|---|---|---|---|---|---|
| I | −1.13891 | −1.418 | 0.0881 ** | 0.035 | 0.001 |
| II | −1.32068 | −19.847 | 0.0506 | 0.033 | 0.002 |
| IIb1 | −1.15945 | −0.649 | 0.2494 | 0.427 | 0.012 |
| IIb2 | 0.00646 | −1.120 | 0.1459 | 0.188 | 0.043 *** |
| IIb3 | −1.96737 * | −15.870 * | 0.0397 ** | 0.014 | 0.001 |
| All | −0.77779 | −13.991 | 0.0696 | 0.006 | 0.010 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ualiyeva, D.; Liu, J.; Dujsebayeva, T.; Li, J.; Tian, L.; Cai, B.; Zeng, X.; Guo, X. Genetic Structure and Population History of the Zaisan Toad-Headed Agama (Phrynocephalus melanurus) Inferred from Mitochondrial DNA. Animals 2024, 14, 209. https://doi.org/10.3390/ani14020209
Ualiyeva D, Liu J, Dujsebayeva T, Li J, Tian L, Cai B, Zeng X, Guo X. Genetic Structure and Population History of the Zaisan Toad-Headed Agama (Phrynocephalus melanurus) Inferred from Mitochondrial DNA. Animals. 2024; 14(2):209. https://doi.org/10.3390/ani14020209
Chicago/Turabian StyleUaliyeva, Daniya, Jinlong Liu, Tatjana Dujsebayeva, Jun Li, Lili Tian, Bo Cai, Xiaomao Zeng, and Xianguang Guo. 2024. "Genetic Structure and Population History of the Zaisan Toad-Headed Agama (Phrynocephalus melanurus) Inferred from Mitochondrial DNA" Animals 14, no. 2: 209. https://doi.org/10.3390/ani14020209
APA StyleUaliyeva, D., Liu, J., Dujsebayeva, T., Li, J., Tian, L., Cai, B., Zeng, X., & Guo, X. (2024). Genetic Structure and Population History of the Zaisan Toad-Headed Agama (Phrynocephalus melanurus) Inferred from Mitochondrial DNA. Animals, 14(2), 209. https://doi.org/10.3390/ani14020209