
Multiple Tissues Transcriptome of Zig-Zag Eel (Mastacembelus armatus) with Different Growth Rates

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
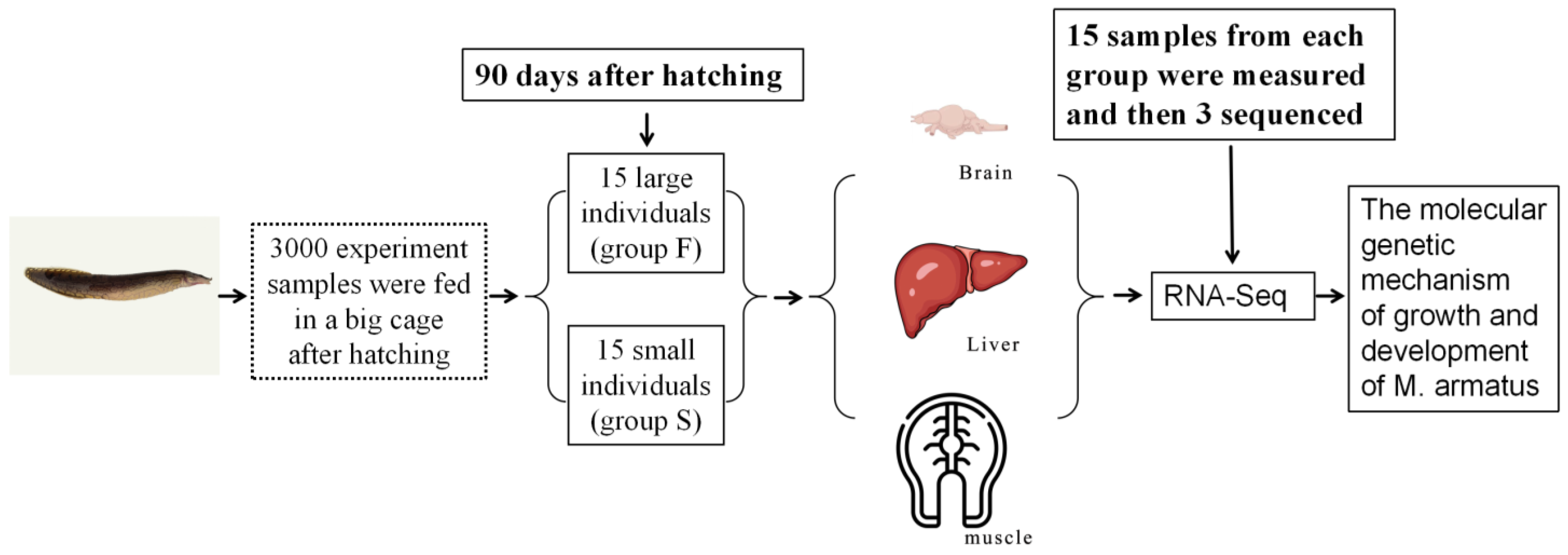
2.1. Fish Maintenance and Specification Measurement
2.2. RNA Extraction, Library Construction, and Transcriptome Sequencing RNA
2.3. Filtering of Clean Reads and Alignment Clean Reads
2.4. DEGs and Enrichment Analysis
2.5. Principal Component Analysis
2.6. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) Validation
2.7. Statistical Analysis
3. Results
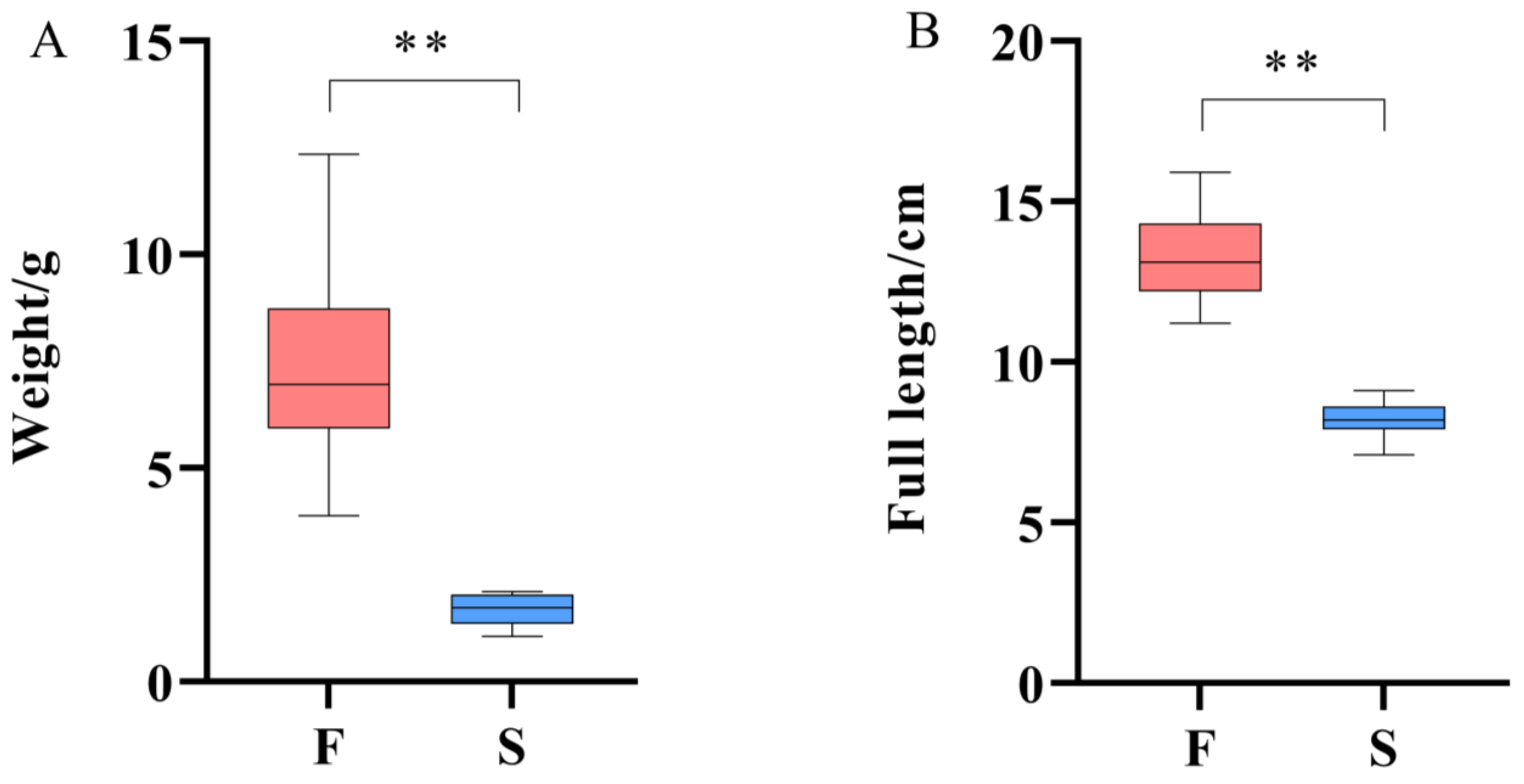
3.1. Analysis of Growth Data
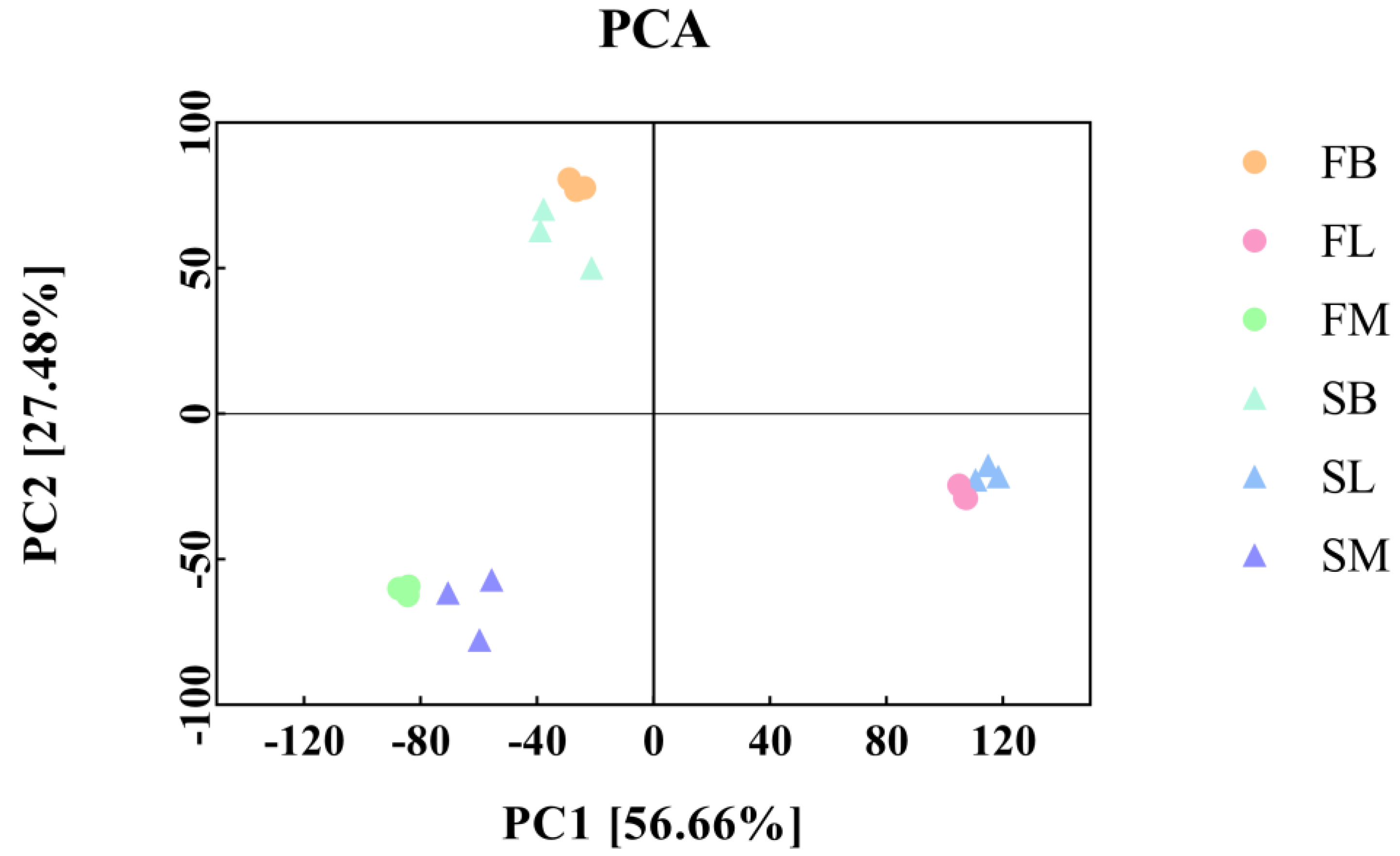
3.2. Transcriptome Sequencing Data, Quality Processing, and Principal Component Analysis
3.3. Screening and Verification of DEGs
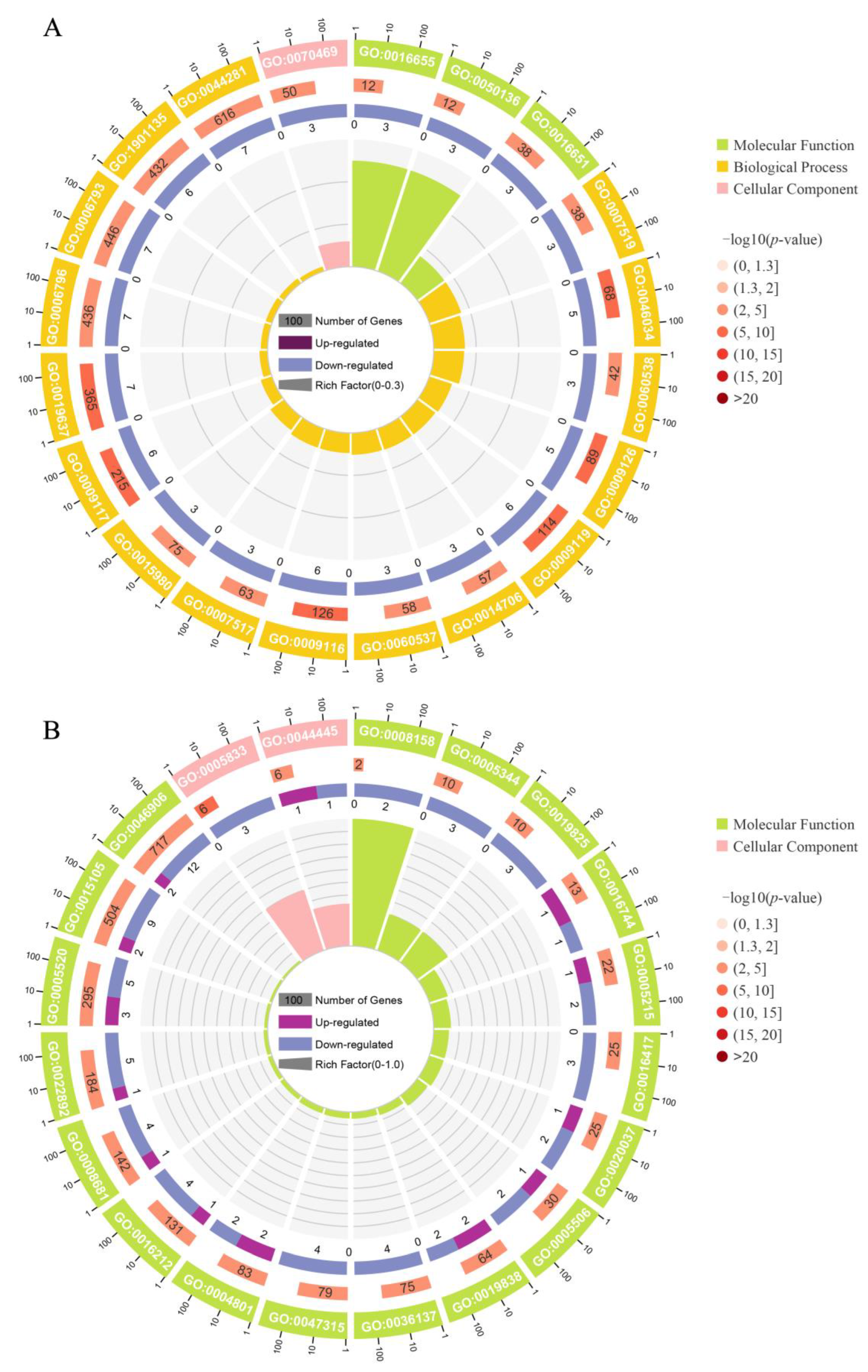
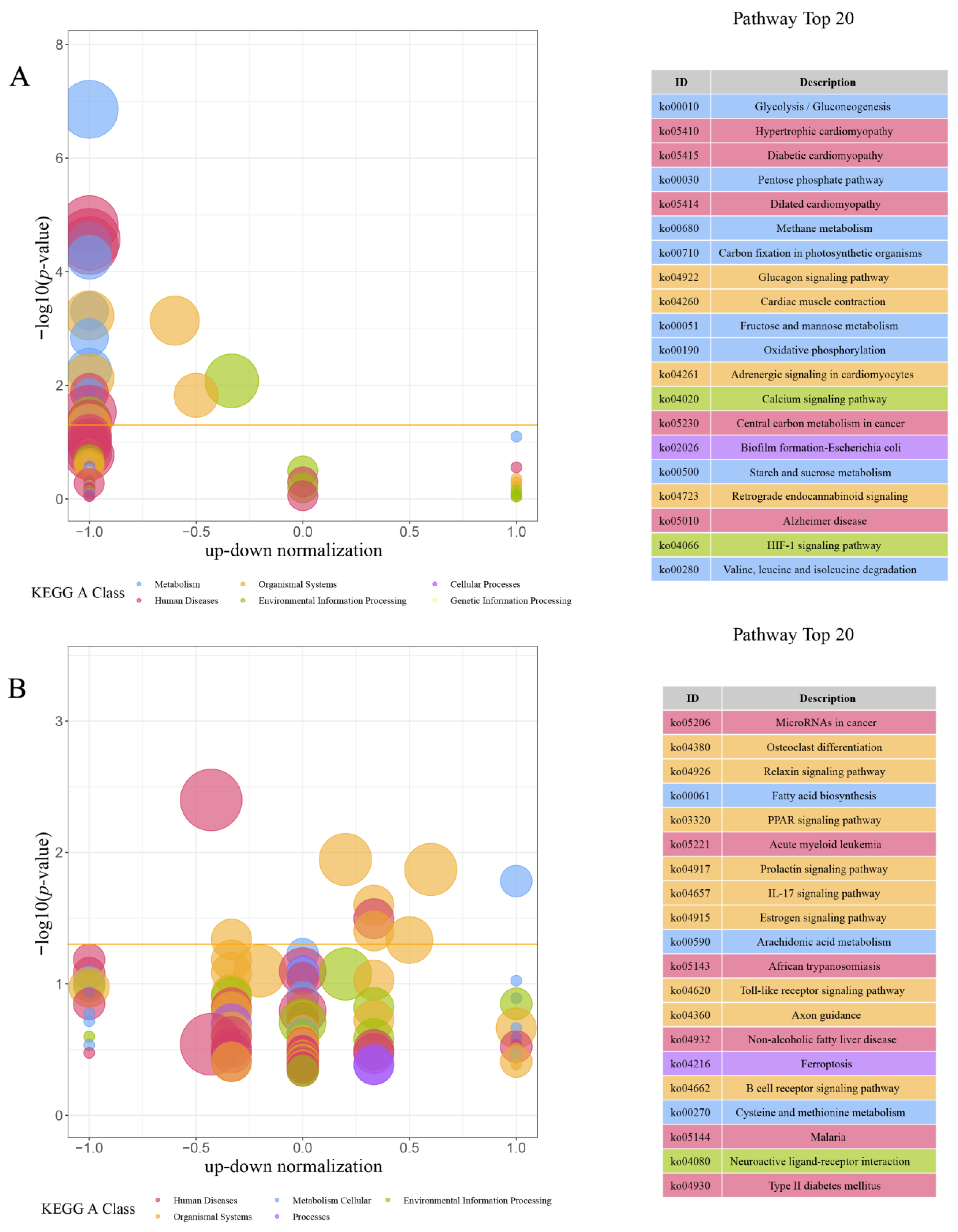
3.4. GO and KEGG Enrichment Analysis
3.5. Identification of Differentially Expressed Growth Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adamek, D.; Rzepkowska, M.; Panagiotopoulou, H.; Ostaszewska, T.; Fajkowska, M.; Kamaszewski, M.; Kolman, R. Morphological Differences of White Muscle Fibers and Genetic Diversity of Fast and Slow Growing Atlantic Sturgeons (Acipenser oxyrinchus). Turk. J. Fish. Aquat. Sci. 2017, 17, 959–966. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, Z.; Solberg, M.F.; Chen, Z.; Wei, M.; Zhu, F.; Jia, C.; Meng, Q.; Zhang, Z. Comparative transcriptome analysis of mixed tissues of black porgy (Acanthopagrus schlegelii) with differing growth rates. Aquac. Res. 2021, 52, 5800–5813. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, C.Y.; Wang, D.D.; Li, X.F.; Xiao, L.; Zhang, X.; You, X.; Shi, Q.; Hu, G.J.; Fang, C.; et al. Transcriptome analysis reveals the molecular mechanisms underlying growth superiority in a novel grouper hybrid (Epinephelus fuscogutatusfemale symbol × E. lanceolatusmale symbol). BMC Genet. 2016, 17, 24. [Google Scholar] [CrossRef]
- Valente, L.M.P.; Moutou, K.A.; Conceicao, L.E.C.; Engrola, S.; Fernandes, J.M.O.; Johnston, L.A. What determines growth potential and juvenile quality of farmed fish species? Rev. Aquac. 2013, 5, S168–S193. [Google Scholar] [CrossRef]
- Tamayo, D.; Ibarrola, I.; Urrutia, M.B.; Navarro, E. The physiological basis for inter-individual growth variability in the spat of clams (Ruditapes philippinarum). Aquaculture 2011, 321, 113–120. [Google Scholar] [CrossRef]
- Liao, X.; Cheng, L.; Xu, P.; Lu, G.; Wachholtz, M.; Sun, X.; Chen, S. Transcriptome analysis of crucian carp (Carassius auratus), an important aquaculture and hypoxia-tolerant species. PLoS ONE 2013, 8, e62308. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, Y.; Xu, X.; Dai, Y.; Li, J. Transcriptome Analysis of the Liver and Muscle Tissues of Black Carp (Mylopharyngodon piceus) of Different Growth Rates. Mar. Biotechnol. 2020, 22, 706–716. [Google Scholar] [CrossRef]
- Fu, B.; Yu, X.; Tong, J.; Pang, M.; Zhou, Y.; Liu, Q.; Tao, W. Comparative transcriptomic analysis of hypothalamus-pituitary-liver axis in bighead carp (Hypophthalmichthys nobilis) with differential growth rate. BMC Genom. 2019, 20, 328. [Google Scholar] [CrossRef]
- Fan, H.; Qiu, M.; Zhong, Q.; Xue, L.; Qin, Z. Comparison of nutritional components of wild and farmed Mastacembelus armatus at different growth stages. J. Anhui Agric. Sci. 2018, 46, 92–96. [Google Scholar]
- Zhong, D.; Huang, X.; He, P.; Zhou, H.; Zhang, M.; Shu, H. Cloning and expression analysis of growth hormone gene of Mastacembelus armatus. J. South. Agric. 2020, 51, 194–201. [Google Scholar]
- Zhong, D.; Lai, X.; Zhang, M.; Shu, H. Cloning and expression analysis of mstn gene of Mastacembelus armatus. J. Guangdong Ocean Univ. 2020, 40, 14–21. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Xue, L.; Gao, Y.; Wu, M.; Tian, T.; Fan, H.; Huang, Y.; Huang, Z. Telomere-to-telomere assembly of a fish Y chromosome reveals the origin of a young sex chromosome pair. Genome Biol. 2021, 22, 203. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Wang, K.; Bu, T.; Cheng, Q.; Dong, L.; Su, T.; Chen, Z.; Kong, F.; Gong, Z.; Liu, B.; Li, M. Two homologous LHY pairs negatively control soybean drought tolerance by repressing the abscisic acid responses. New Phytol. 2021, 229, 2660–2675. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Chapman, R.W.; Reading, B.J.; Sullivan, C.V. Ovary transcriptome profiling via artificial intelligence reveals a transcriptomic fingerprint predicting egg quality in striped bass, Morone saxatilis. PLoS ONE 2014, 9, e96818. [Google Scholar] [CrossRef]
- Gudbrandsson, J.; Ahi, E.P.; Franzdottir, S.R.; Kapralova, K.H.; Kristjansson, B.K.; Steinhaeuser, S.S.; Maier, V.H.; Johannesson, I.M.; Snorrason, S.S.; Jonsson, Z.O.; et al. The developmental transcriptome of contrasting Arctic charr (Salvelinus alpinus) morphs. F1000Research 2015, 4, 136. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, F.; Huang, J.; Yang, L.; Jiang, S.; Yan, Q.; Jiang, S. Transcriptome and miRNA Profiles of Black Tiger Shrimp, Penaeus monodon, Under Different Salinity Conditions. Front. Mar. Sci. 2020, 7, 579381. [Google Scholar] [CrossRef]
- Jeukens, J.; Renaut, S.; St-cyr, J.; Nolte, A.W.; Bernatchez, L. The transcriptomics of sympatric dwarf and normal lake whitefish (Coregonus clupeaformis spp., Salmonidae) divergence as revealed by next-generation sequencing. Mol. Ecol. 2010, 19, 5389–5403. [Google Scholar] [CrossRef]
- Wang, R.; Hu, X.; Lü, A.; Liu, R.; Sun, J.; Sung, Y.Y.; Song, Y. Transcriptome analysis in the skin of Carassius auratus challenged with Aeromonas hydrophila. Fish Shellfish Immunol. 2019, 94, 510–516. [Google Scholar] [CrossRef]
- Guan, W.Z.; Qiu, G.F.; Liu, F. Transcriptome analysis of the growth performance of hybrid mandarin fish after food conversion. PLoS ONE 2020, 15, e0240308. [Google Scholar] [CrossRef]
- Kim, H.; Barton, E.; Muja, N.; Yakar, S.; Pennisi, P.; LeRoith, D. Intact Insulin and Insulin-Like Growth Factor-I Receptor Signaling Is Required for Growth Hormone Effects on Skeletal Muscle Growth and Function in Vivo. Endocrinology 2005, 146, 1772–1779. [Google Scholar] [CrossRef]
- Ndandala, C.B.; Dai, M.; Mustapha, U.F.; Li, X.; Liu, J.; Huang, H.; Li, G.; Chen, H. Current research and future perspectives of GH and IGFs family genes in somatic growth and reproduction of teleost fish. Aquac. Rep. 2022, 26, 101289. [Google Scholar] [CrossRef]
- Triantaphyllopoulos, K.A.; Cartas, D.; Miliou, H. Factors influencing GH and IGF-I gene expression on growth in teleost fish: How can aquaculture industry benefit? Rev. Aquac. 2019, 12, 1637–1662. [Google Scholar] [CrossRef]
- Roith, D.L.; Bondy, C.; Yakar, S.; Liu, J.L.; Butler, A. The Somatomedin Hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74. [Google Scholar] [CrossRef]
- Pérez-Sánchez, J. The involvement of growth hormone in growth regulation, energy homeostasis and immune function in the gilthead sea bream (Sparus aurata): A short review. Fish Physiol. Biochem. 2000, 22, 135–144. [Google Scholar] [CrossRef]
- Sjögren, K.; Liu, J.L.; Blad, K.; Skrtic, S.; Vidal, O.; Wallenius, V.; Leroith, D.; Törnell, J.; Isaksson, O.G.P.; Jansson, J.O.; et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7088–7092. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Liu, J.L.; Stannard, B.; Butler, A.; Accili, D.; Sauer, B.; Leroith, D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1999, 96, 7324–7329. [Google Scholar] [CrossRef]
- Funkenstein, B.; Almuly, R.; Chan, S.J. Localization of IGF-I and IGF-I Receptor mRNA inSparus aurataLarvae. Gen. Comp. Endocrinol. 1997, 107, 291–303. [Google Scholar] [CrossRef]
- Silva, A.C.G.; Almeida, D.V.; Nornberg, B.F.; Pereira, J.R.; Pires, D.M.; Corcini, C.D.; Junior, A.S.V.; Marins, L.F. Reproductive parameters of double transgenic zebrafish (Danio rerio) males overexpressing both the growth hormone (GH) and its receptor (GHR). Transgenic Res. 2017, 26, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Daughaday, W.H.; Rotwein, P. Insulin-Like Growth Factors I and II. Peptide, Messenger Ribonucleic Acid and Gene Structures, Serum, and Tissue Concentrations. Endocr. Rev. 1989, 10, 68–91. [Google Scholar] [CrossRef]
- Kamei, H.; Lu, L.; Jiao, S.; Li, Y.; Gyrup, C.; Laursen, L.S.; Oxvig, C.; Zhou, J.; Duan, C. Duplication and diversification of the hypoxia-inducible IGFBP-1 gene in zebrafish. PLoS ONE 2008, 3, e3091. [Google Scholar] [CrossRef] [PubMed]
- Wheatcroft, S.B.; Kearney, M.T. IGF-dependent and IGF-independent actions of IGF-binding protein-1 and -2: Implications for metabolic homeostasis. Trends Endocrinol. Metab. 2009, 20, 153–162. [Google Scholar] [CrossRef]
- Tanaka, H.; Oishi, G.; Nakano, Y.; Mizuta, H.; Nagano, Y.; Hiramatsu, N.; Ando, H.; Shimizu, M. Production of recombinant salmon insulin-like growth factor binding protein-1 subtypes. Gen. Comp. Endocrinol. 2018, 257, 184–191. [Google Scholar] [CrossRef]
- Fruchtman, S.; Mcvey, D.C.; Borski, R.J. Characterization of pituitary IGF-I receptors: Modulation of prolactin and growth hormone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R468–R476. [Google Scholar] [CrossRef]
- Wittenberg, J.B.; Wittenberg, B.A. Myoglobin function reassessed. J. Exp. Biol. 2003, 206, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Ueki, N.; Ochiai, Y. Primary structure and thermostability of bigeye tuna myoglobin in relation to those of other scombridae fish. Fish. Sci. 2004, 70, 875–884. [Google Scholar] [CrossRef]
- Qi, D.; Chao, Y.; Zhao, Y.; Xia, M.; Wu, R. Molecular evolution of myoglobin in the Tibetan Plateau endemic schizothoracine fish (Cyprinidae, Teleostei) and tissue-specific expression changes under hypoxia. Fish Physiol. Biochem. 2017, 44, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Sun, B.; Sun, N.; Li, C.; Zhang, J.; Yang, W. Structure, functional properties and iron bioavailability of Pneumatophorus japonicus myoglobin and its glycosylation products. Int. J. Biol. Macromol. 2021, 173, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Ushio, H.; Ochiai, Y. Expression and characterization of rainbow trout Oncorhynchus mykiss recombinant myoglobin. Int. J. Biol. Macromol. 2021, 47, 1477–1488. [Google Scholar] [CrossRef]
- Nakamura, Y.-N.; Ando, M.; Seoka, M.; Kawasaki, K.; Tsukamasa, Y. Changes of proximate compositions and myoglobin content in the dorsal ordinary muscles of the cultured Pacific bluefin tuna Thunnus orientalis with growth. Fish. Sci. 2007, 73, 1155–1159. [Google Scholar] [CrossRef]
- Lu, X.; Chen, H.M.; Qian, X.Q.; Gui, J.F. Transcriptome analysis of grass carp (Ctenopharyngodon idella) between fast- and slow-growing fish. Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 35, 100688. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′→3′) | Melting Temperature/°C |
|---|---|---|
| ACTA1 | F: GCACCACACCTTCTACAA | 49.5 |
| R: AGCATCCAGCACAATACC | 49.7 | |
| MYL1 | F: GACCTGTCCTCCATCAAG | 49 |
| R: AGCCATTCTCATCCTCCT | 49.3 | |
| ZIC5 | F: GCACGAACTGGTCAATCA | 50.5 |
| R: TGTGAGTCCTCTTGTGAATC | 50.4 | |
| FBP2 | F: GCCATCTACAAGCGGTTAT | 50.5 |
| R: GGTCCAGCATGAAGAAGTT | 50.5 | |
| GDF10 | F: TCCAGCCAACCTACCATAT | 50.2 |
| R: CCTTCTTCATCGTCCTCTC | 49.9 | |
| DUSP2 | F: GGTGGCAGATGAGAACAG | 50.2 |
| R: AGTCAATGAAGGCGATGG | 49.8 | |
| IGF2BP1 | F: CAGACGCAGAGCAAGATT | 50.2 |
| R: AAGTTGTTGTGAGCCAGAA | 50.2 | |
| IGFBP1A | F: AGAGCCTGAGATGGAGAAC | 51.1 |
| R: CCAGAGACGACTCACACT | 50.7 | |
| IGFBP1B | F: CAGCACTGGACACGATAG | 50.1 |
| R: TGACTTCTTGATGACACTCT | 49.3 | |
| ME1 | F: TCACGAGAAGGAGAGGTT | 49.3 |
| R: GGAAGATGTAGGCATTGTTC | 49.8 | |
| ACLYA | F: GGTATGTCCAACGAACTCA | 50 |
| R: TCTGTGCCTCCAATCTCT | 49.6 | |
| MYBPC3 | F: GCCTCCTGTCCTGATTGTGA | 55.0 |
| R: AGCCATAGATTCTCCGCCATT | 55.1 | |
| SDS | F: TGCTGAATGGAGTGGTGGAG | 55.3 |
| R: CCTGGTCTGTGACTACTTCTGA | 54.9 | |
| HAUS5 | F: ATCCATAACAGAGCCACAAGTC | 54.2 |
| R: ACATCCAGAGCAGCCAGTT | 54.2 | |
| TACC3 | F: ACTGAGAACCTGCCGAACAA | 55.1 |
| R: AAGGACTCCATTGCTGCTGTA | 55.2 | |
| CCNB3 | F: TCTCAGTGTCTGTCAGGTTGT | 54.4 |
| R: GAGTTCTCAGCGTCAATGTCA | 54.3 |
| Sample | Clean Paired Reads | Clean Bases (G) | Q20 (%) | Q30 (%) | GC Content (%) | Clean Data Ratio (%) |
|---|---|---|---|---|---|---|
| SB-1 | 52,431,126 | 7.63 | 98.89 | 95.63 | 46.97 | 89.03 |
| SB-2 | 52,766,502 | 7.7 | 98.94 | 95.81 | 47.4 | 89.36 |
| SB-3 | 58,687,688 | 8.55 | 98.86 | 95.53 | 47.29 | 89.32 |
| SL-1 | 43,273,446 | 6.32 | 98.96 | 95.81 | 48.69 | 90.69 |
| SL-2 | 50,526,274 | 7.37 | 99.02 | 96.03 | 49.28 | 90.26 |
| SL-3 | 46,867,612 | 6.85 | 99.01 | 96.01 | 49.01 | 90.15 |
| SM-1 | 45,096,312 | 6.61 | 98.97 | 95.86 | 50.44 | 90.3 |
| SM-2 | 53,343,670 | 7.8 | 98.99 | 95.98 | 49.89 | 90.07 |
| SM-3 | 47,723,246 | 6.99 | 99.06 | 96.19 | 50.41 | 90.64 |
| FB-1 | 37,071,366 | 5.45 | 98.77 | 95.22 | 46.61 | 90.16 |
| FB-2 | 36,020,462 | 5.3 | 98.74 | 95.14 | 46.38 | 89.86 |
| FB-3 | 40,536,446 | 5.97 | 98.75 | 95.18 | 46.91 | 89.6 |
| FL-1 | 40,133,186 | 5.87 | 98.89 | 95.62 | 48.61 | 89.05 |
| FL-2 | 41,425,186 | 6.04 | 99.06 | 96.17 | 49.01 | 89.94 |
| FL-3 | 43,314,210 | 6.4 | 98.81 | 95.34 | 48.48 | 89.45 |
| FM-1 | 39,622,272 | 5.82 | 98.97 | 95.89 | 50.94 | 90.27 |
| FM-2 | 40,047,894 | 5.85 | 99.02 | 96.03 | 50.78 | 89.99 |
| FM-3 | 45,607,112 | 6.68 | 98.95 | 95.83 | 50.73 | 90.12 |
| Sample | Total Reads | Paired Mapped Reads (%) | Unpaired Mapped Reads (%) | Unmapped Reads (%) | Total Mapped Reads (%) |
|---|---|---|---|---|---|
| SB-1 | 51,517,494 | 38,344,392 (74.43%) | 3,161,940 (6.14%) | 10,011,162 (19.43%) | 41,506,332 (80.57%) |
| SB-2 | 51,613,212 | 36,682,906 (71.07%) | 3,175,401 (6.15%) | 11,754,905 (22.77%) | 39,858,307 (77.23%) |
| SB-3 | 57,178,028 | 40,208,324 (70.32%) | 3,507,018 (6.13%) | 13,462,686 (23.55%) | 43,715,342 (76.45%) |
| SL-1 | 42,519,140 | 30,457,448 (71.63%) | 2,904,978 (6.83%) | 9,156,714 (21.54%) | 33,362,426 (78.46%) |
| SL-2 | 49,550,142 | 35,433,480 (71.51%) | 3,105,560 (6.27%) | 11,011,102 (22.22%) | 38,539,040 (77.78%) |
| SL-3 | 45,301,398 | 31,087,930 (68.62%) | 3,084,443 (6.81%) | 11,129,025 (24.57%) | 34,172,373 (75.43%) |
| SM-1 | 44,805,510 | 36,928,266 (82.42%) | 2,044,978 (4.56%) | 5,832,266 (13.02%) | 38,973,244 (86.98%) |
| SM-2 | 53,099,480 | 40,588,834 (76.44%) | 3,085,672 (5.81%) | 9,424,974 (17.75%) | 43,674,506 (82.25%) |
| SM-3 | 47,381,560 | 38,406,404 (81.06%) | 2,163,296 (4.57%) | 6,811,860 (14.38%) | 40,569,700 (85.62%) |
| FB-1 | 36,626,502 | 25,861,926 (70.61%) | 2,666,815 (7.28%) | 8,097,761 (22.11%) | 28,528,741 (77.89%) |
| FB-2 | 35,652,742 | 24,567,788 (68.91%) | 2,849,139 (7.99%) | 8,235,815 (23.10%) | 27,416,927 (76.90%) |
| FB-3 | 39,921,844 | 28,220,434 (70.69%) | 2,825,609 (7.08%) | 8,875,801 (22.23%) | 31,046,043 (77.77%) |
| FL-1 | 39,241,700 | 26,444,650 (67.39%) | 2,679,001 (6.83%) | 10,118,049 (25.78%) | 29,123,651 (74.22%) |
| FL-2 | 40,425,420 | 27,880,806 (68.97%) | 2,630,530 (6.51%) | 9,914,084 (24.52%) | 30,511,336 (75.48%) |
| FL-3 | 42,839,680 | 28,279,204 (66.01%) | 3,870,241 (9.03%) | 10,690,235 (24.95%) | 32,149,445 (75.05%) |
| FM-1 | 38,794,238 | 32,069,580 (82.67%) | 1,665,771 (4.29%) | 5,058,887 (13.04%) | 33,735,351 (86.96%) |
| FM-2 | 39,748,678 | 33,003,178 (83.03%) | 1,608,890 (4.05%) | 5,136,610 (12.92%) | 34,612,068 (87.08%) |
| FM-3 | 45,296,212 | 37,355,274 (82.47%) | 2,009,298 (4.44%) | 5,931,640 (13.10%) | 39,364,572 (86.90%) |
| ID | Log2FC (FDR < 0.05) | Gene | Description |
|---|---|---|---|
| 113122805 | −2.822629123 | ACTA1B | Actin alpha 1, skeletal muscle b. |
| 113141556 | −2.92542818 | MYLPFA | Myosin light chain, phosphorylatable, fast skeletal muscle a. |
| 113123291 | −2.913046983 | MYL1 | Myosin, light chain 1, alkali; skeletal, fast. |
| 113132910 | −3.131707241 | TNNT3A | Troponin T type 3a (skeletal, fast). |
| 113127528 | −3.002337731 | MYLZ3 | Myosin, light polypeptide 3, skeletal muscle. |
| 113135302 | −2.965010599 | LDB3B | LIM domain binding 3b. |
| 113135777 | −2.476991992 | ACTN3B | Actinin alpha 3b. |
| 113123100 | 2.966333471 | ZIC5 | Zic family member 5. |
| 113132015 | −2.568776028 | SYNPO2LA | Synaptopodin 2-like a. |
| ID | Log2FC (FDR < 0.05) | Gene | Description |
|---|---|---|---|
| 113141522 | −3.854791781 | IGF2BP1 | Insulin-like growth factor 2 mRNA binding protein 1. |
| 113139767 | −3.682401004 | IGFBP1A | Insulin-like growth factor binding protein 1a. |
| 113134287 | −2.810482959 | IGFBP1B | Insulin-like growth factor binding protein 1b. |
| 113130337 | −2.274576177 | ITGA7 | Integrin, alpha 7. |
| 113122938 | −2.001084859 | IRS2B | Insulin receptor substrate 2b |
| 113141142 | 2.623058494 | ACLYA | ATP citrate lyase a. |
| 113130328 | 2.160598057 | MYL9B | Myosin, light chain 9b, regulatory. |
| ID | Log2FC (FDR < 0.05) | Gene | Description |
|---|---|---|---|
| 113130167 | −3.846745356 | CDKN1A | Cyclin-dependent kinase inhibitor 1A. |
| 113134287 | −3.870604985 | IGFBP1B | Insulin-like growth factor binding protein 1b. |
| 113141522 | −2.699824093 | IGF2BP1 | Insulin-like growth factor 2 mRNA binding protein 1. |
| 113126273 | −2.520499238 | IGF2BP3 | Insulin-like growth factor 2 mRNA binding protein 3. |
| 113136432 | 2.379825043 | MYOM3 | Myomesin 3. |
| 113128157 | 2.737550286 | MB | Myoglobin. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Lu, B.; Yu, Z.; Zhang, L.; Chen, Y.; Chen, Z.; Han, C.; Shu, H. Multiple Tissues Transcriptome of Zig-Zag Eel (Mastacembelus armatus) with Different Growth Rates. Animals 2024, 14, 248. https://doi.org/10.3390/ani14020248
Yang J, Lu B, Yu Z, Zhang L, Chen Y, Chen Z, Han C, Shu H. Multiple Tissues Transcriptome of Zig-Zag Eel (Mastacembelus armatus) with Different Growth Rates. Animals. 2024; 14(2):248. https://doi.org/10.3390/ani14020248
Chicago/Turabian StyleYang, Jinlin, Baoyue Lu, Zhide Yu, Linan Zhang, Yiman Chen, Zihui Chen, Chong Han, and Hu Shu. 2024. "Multiple Tissues Transcriptome of Zig-Zag Eel (Mastacembelus armatus) with Different Growth Rates" Animals 14, no. 2: 248. https://doi.org/10.3390/ani14020248
APA StyleYang, J., Lu, B., Yu, Z., Zhang, L., Chen, Y., Chen, Z., Han, C., & Shu, H. (2024). Multiple Tissues Transcriptome of Zig-Zag Eel (Mastacembelus armatus) with Different Growth Rates. Animals, 14(2), 248. https://doi.org/10.3390/ani14020248