
Improved Equine Fecal Microbiome Characterization Using Target Enrichment by Hybridization Capture
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. 16S rRNA Amplicon Library Preparation
2.3. Target Enrichment by Hybridization Capture (TEHC)
2.4. Sequencing and Data Analysis
3. Results
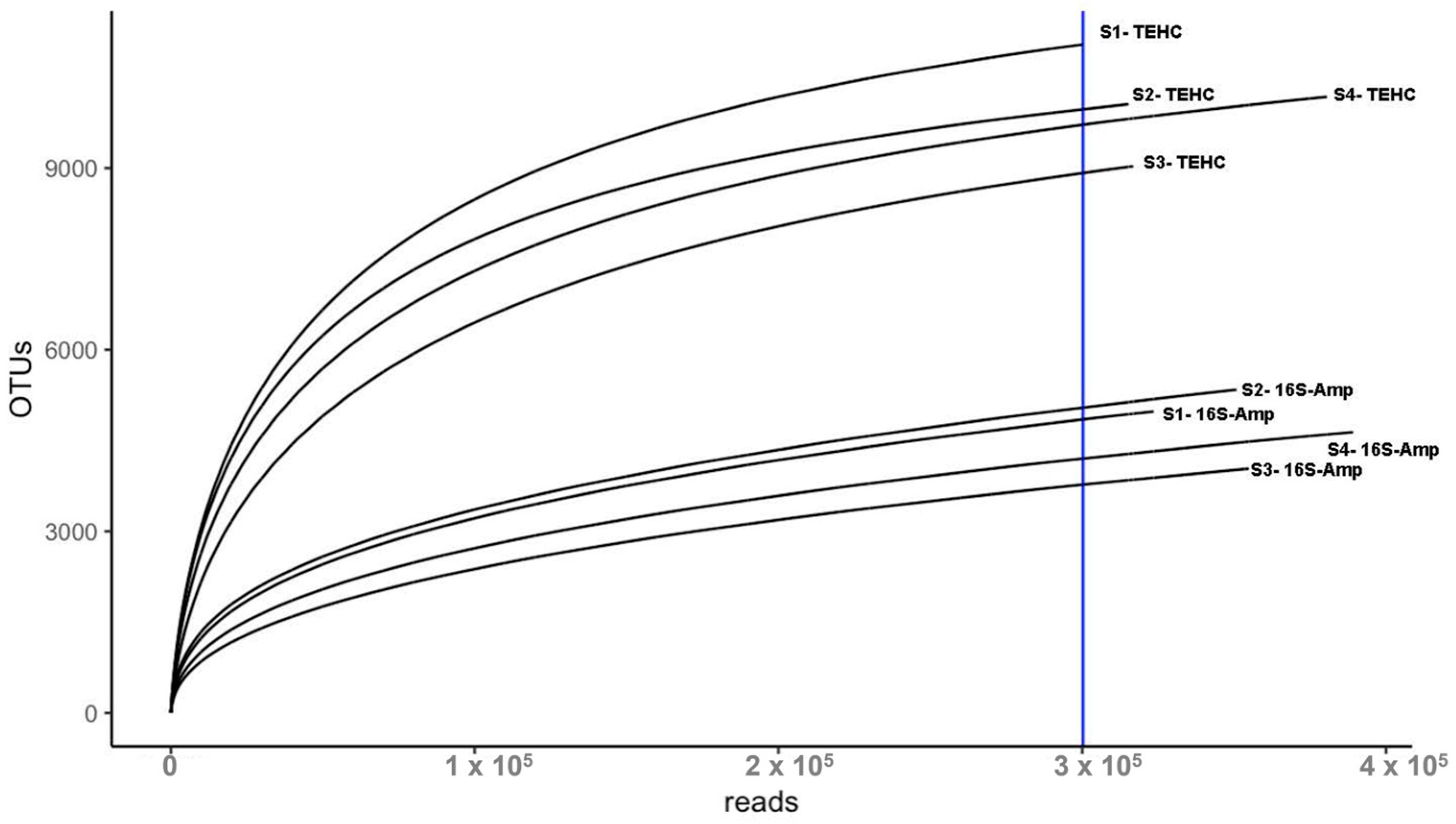
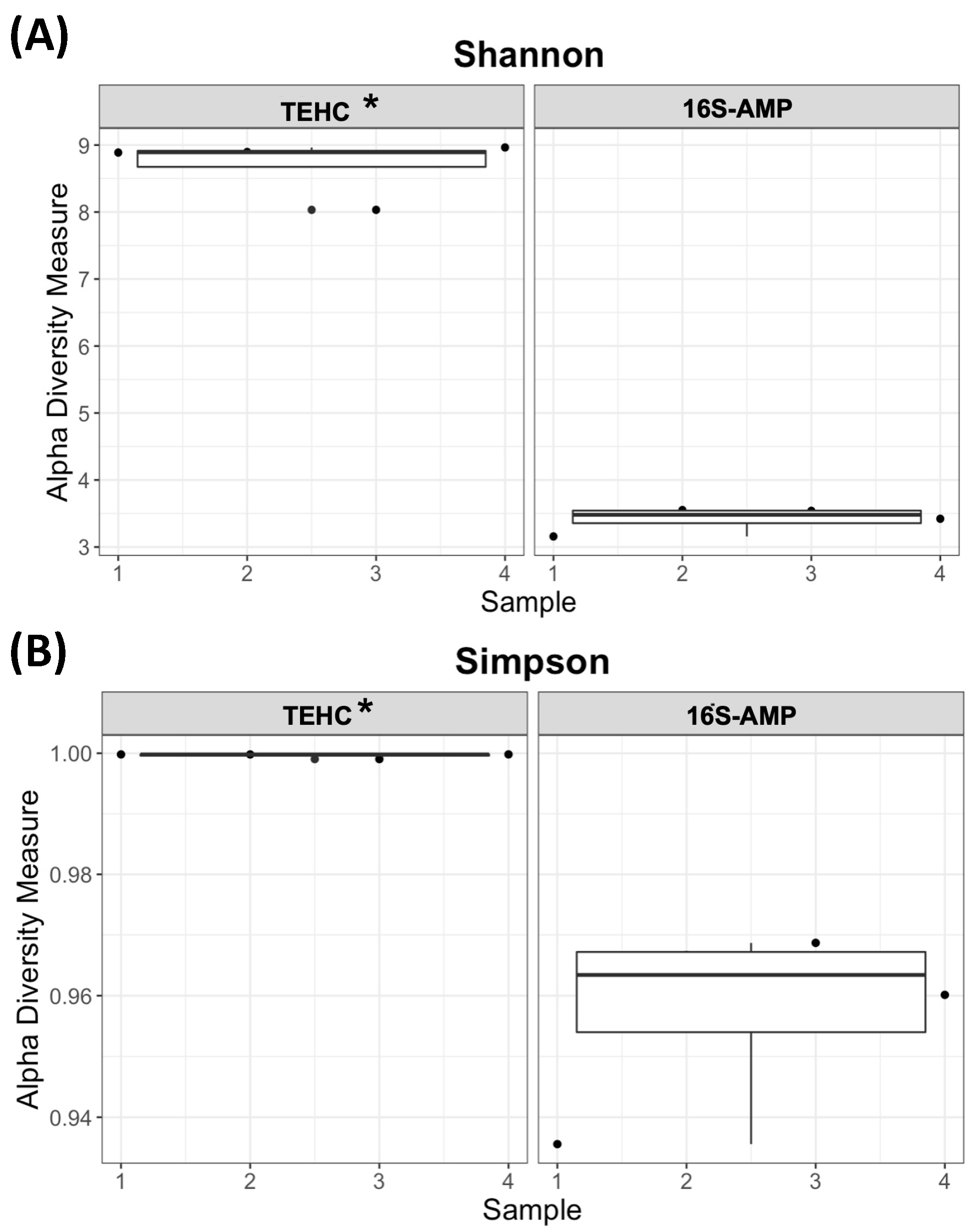
3.1. TEHC Reveals more OTUs Than Conventional 16S Amplicon Sequencing
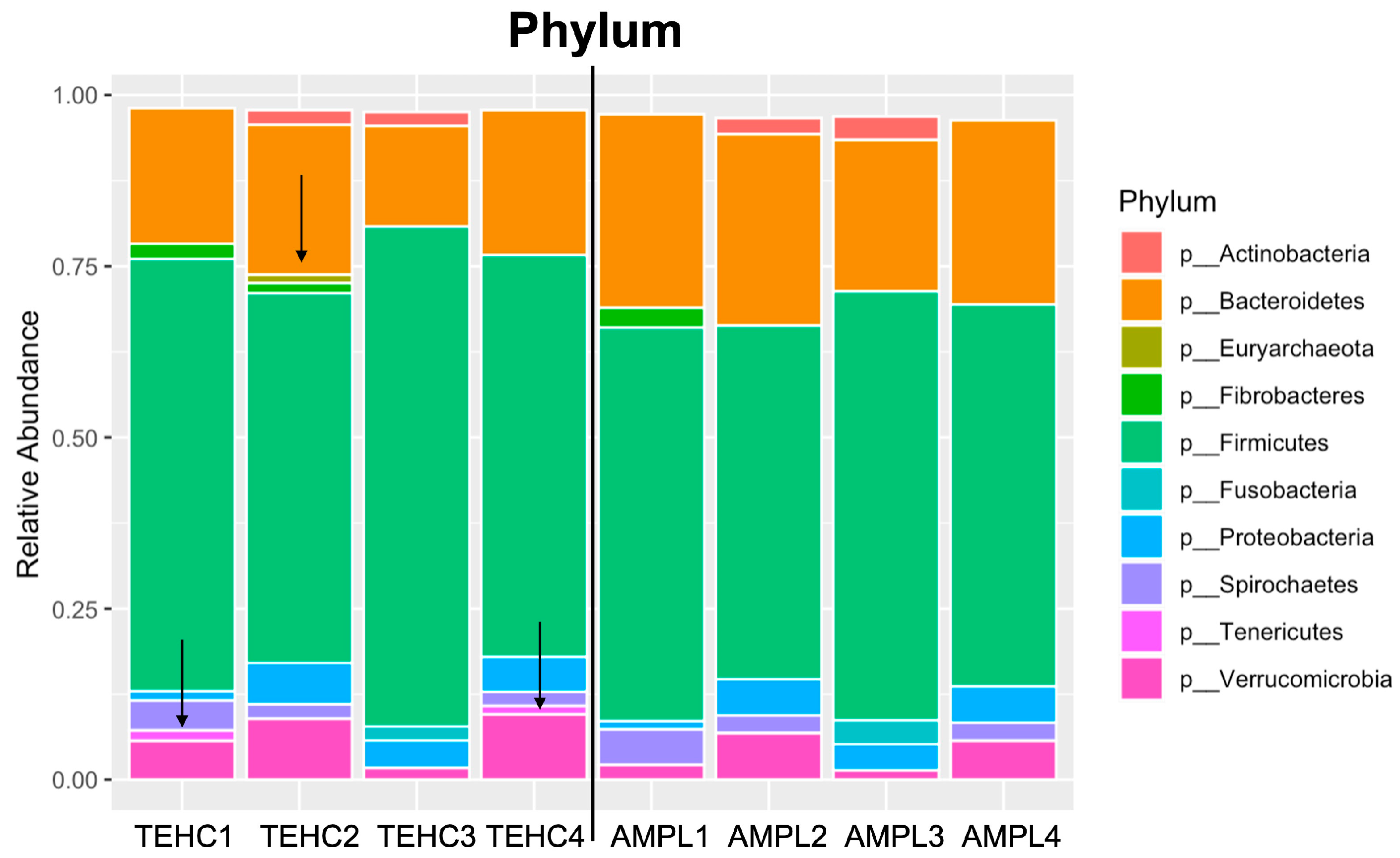
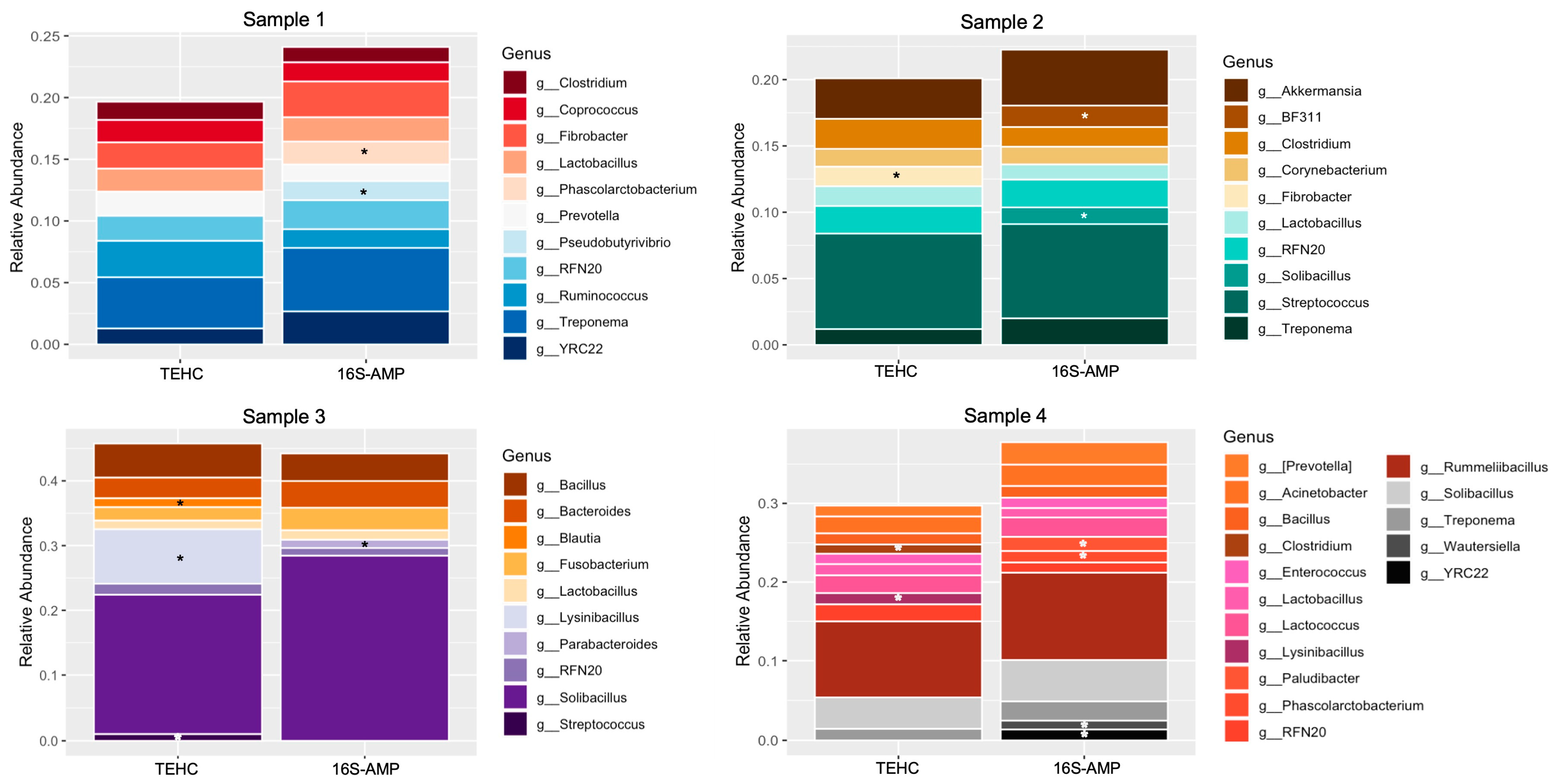
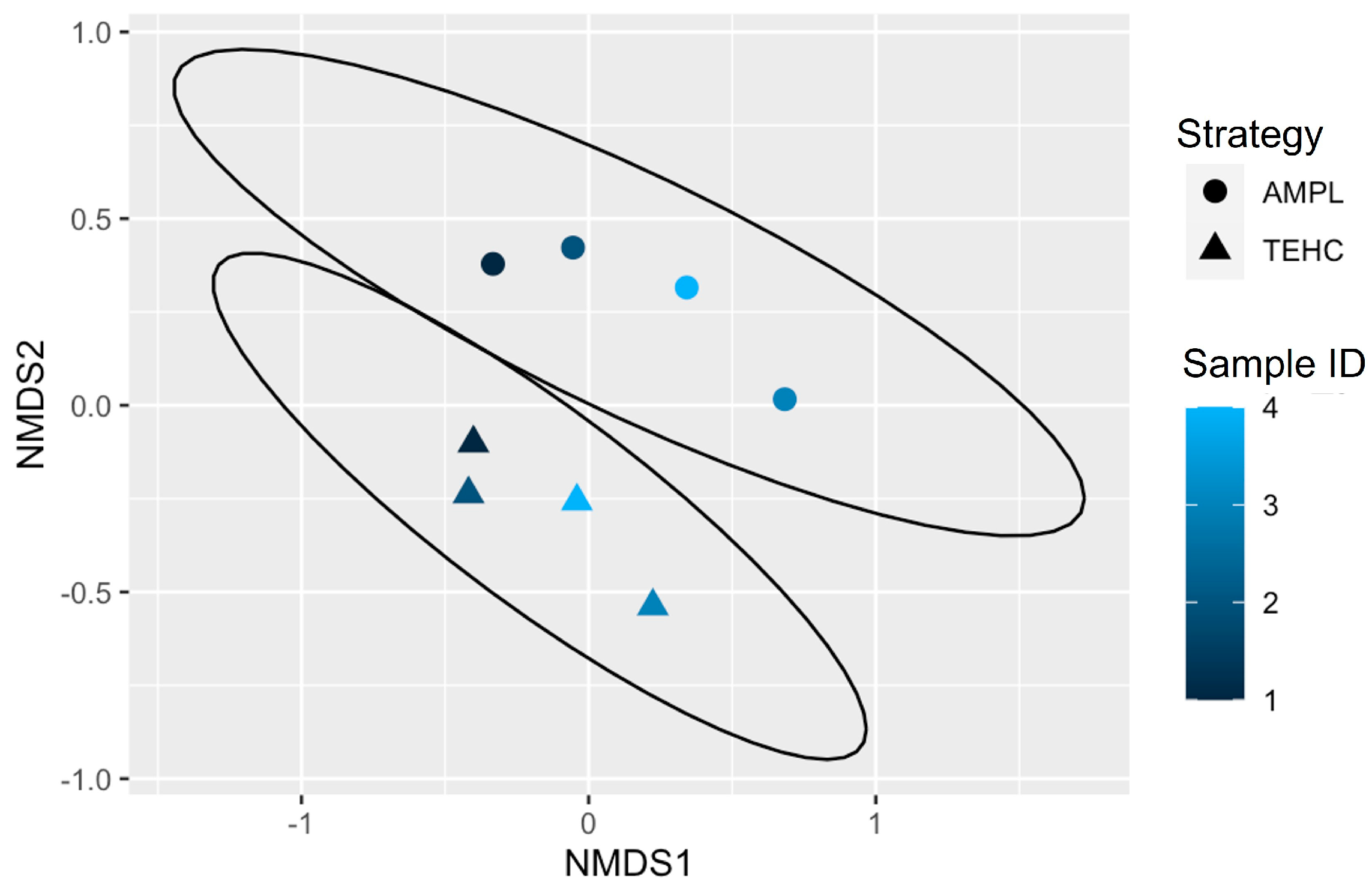
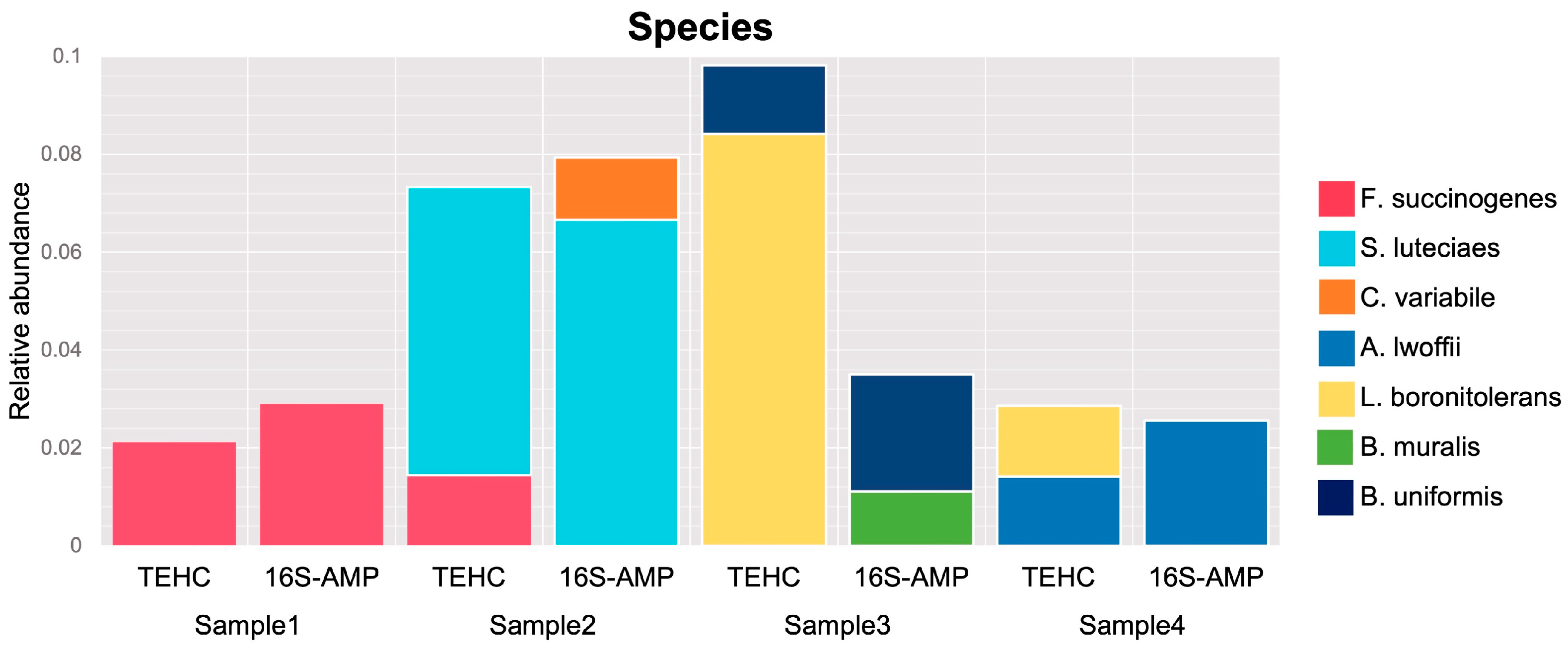
3.2. TEHC-Sequenced Samples Present a Richer and more Diverse Microbial Community Composition Than Those Sequenced following the 16S Amplicon Approach
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- USDA. Equine Mortality in the United States, 2015; USDA, APHIS: Riverdale, MD, USA, 2017.
- Salem, S.E.; Maddox, T.W.; Antczak, P.; Ketley, J.M.; Williams, N.J.; Archer, D.C. Acute changes in the colonic microbiota are associated with large intestinal forms of surgical colic. BMC Vet. Res. 2019, 15, 468. [Google Scholar] [CrossRef]
- Garber, A.; Hastie, P.; Murray, J.A. Factors Influencing Equine Gut Microbiota: Current Knowledge. J. Equine Vet. Sci. 2020, 88, 102943. [Google Scholar] [CrossRef]
- Park, T.; Cheong, H.; Yoon, J.; Kim, A.; Yun, Y.; Unno, T. Comparison of the Fecal Microbiota of Horses with Intestinal Disease and Their Healthy Counterparts. Vet. Sci. 2021, 8, 113. [Google Scholar] [CrossRef]
- Stewart, H.L.; Southwood, L.L.; Indugu, N.; Vecchiarelli, B.; Engiles, J.B.; Pitta, D. Differences in the equine faecal microbiota between horses presenting to a tertiary referral hospital for colic compared with an elective surgical procedure. Equine Vet. J. 2019, 51, 336–342. [Google Scholar] [CrossRef]
- Arnold, C.; Pilla, R.; Chaffin, K.; Lidbury, J.; Steiner, J.; Suchodolski, J. Alterations in the Fecal Microbiome and Metabolome of Horses with Antimicrobial-Associated Diarrhea Compared to Antibiotic-Treated and Non-Treated Healthy Case Controls. Animals 2021, 11, 1807. [Google Scholar] [CrossRef]
- Costa, M.C.; Arroyo, L.G.; Allen-Vercoe, E.; Stampfli, H.R.; Kim, P.T.; Sturgeon, A.; Weese, J.S. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS ONE 2012, 7, e41484. [Google Scholar] [CrossRef]
- McKinney, C.A.; Oliveira, B.C.M.; Bedenice, D.; Paradis, M.-R.; Mazan, M.; Sage, S.; Sanchez, A.; Widmer, G. The fecal microbiota of healthy donor horses and geriatric recipients undergoing fecal microbial transplantation for the treatment of diarrhea. PLoS ONE 2020, 15, e0230148. [Google Scholar] [CrossRef]
- Costa, M.C.; Stampfli, H.R.; Arroyo, L.G.; Allen-Vercoe, E.; Gomes, R.G.; Weese, J.S. Changes in the equine fecal microbiota associated with the use of systemic antimicrobial drugs. BMC Vet. Res. 2015, 11, 19. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Fernandes, K.A.; Kittelmann, S.; Rogers, C.W.; Gee, E.K.; Bolwell, C.F.; Bermingham, E.N.; Thomas, D.G. Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PLoS ONE 2014, 9, e112846. [Google Scholar] [CrossRef]
- Mach, N.; Ruet, A.; Clark, A.; Bars-Cortina, D.; Ramayo-Caldas, Y.; Crisci, E.; Pennarun, S.; Dhorne-Pollet, S.; Foury, A.; Moisan, M.P.; et al. Priming for welfare: Gut microbiota is associated with equitation conditions and behavior in horse athletes. Sci. Rep. 2020, 10, 8311. [Google Scholar] [CrossRef]
- Alvarez-Narvaez, S.; Berghaus, L.J.; Morris, E.R.A.; Willingham-Lane, J.M.; Slovis, N.M.; Giguere, S.; Cohen, N.D. A Common Practice of Widespread Antimicrobial Use in Horse Production Promotes Multi-Drug Resistance. Sci. Rep. 2020, 10, 911. [Google Scholar] [CrossRef]
- Shepherd, M.L.; Swecker, W.S., Jr.; Jensen, R.V.; Ponder, M.A. Characterization of the fecal bacteria communities of forage-fed horses by pyrosequencing of 16S rRNA V4 gene amplicons. FEMS Microbiol. Lett. 2012, 326, 62–68. [Google Scholar] [CrossRef]
- Martinez-Porchas, M.; Villalpando-Canchola, E.; Vargas-Albores, F. Significant loss of sensitivity and specificity in the taxonomic classification occurs when short 16S rRNA gene sequences are used. Heliyon 2016, 2, e00170. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, B.; Bai, D.; Huang, J.; Shiraigo, W.; Yang, L.; Zhao, Q.; Ren, X.; Wu, J.; Bao, W.; et al. Comparison of Fecal Microbiota of Mongolian and Thoroughbred Horses by High-throughput Sequencing of the V4 Region of the 16S rRNA Gene. Asian-Australas. J. Anim. Sci. 2016, 29, 1345–1352. [Google Scholar] [CrossRef]
- Vetrovsky, T.; Baldrian, P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE 2013, 8, e57923. [Google Scholar] [CrossRef]
- Neelakanta, G.; Sultana, H. The use of metagenomic approaches to analyze changes in microbial communities. Microbiol. Insights 2013, 6, 37–48. [Google Scholar] [CrossRef]
- Gilroy, R.; Leng, J.; Ravi, A.; Adriaenssens, E.M.; Oren, A.; Baker, D.; La Ragione, R.M.; Proudman, C.; Pallen, M.J. Metagenomic investigation of the equine faecal microbiome reveals extensive taxonomic diversity. PeerJ 2022, 10, e13084. [Google Scholar] [CrossRef]
- Ang, L.; Vinderola, G.; Endo, A.; Kantanen, J.; Jingfeng, C.; Binetti, A.; Burns, P.; Qingmiao, S.; Suying, D.; Zujiang, Y.; et al. Gut Microbiome Characteristics in feral and domesticated horses from different geographic locations. Commun. Biol. 2022, 5, 172. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef]
- Gasc, C.; Peyret, P. Hybridization capture reveals microbial diversity missed using current profiling methods. Microbiome 2018, 6, 61. [Google Scholar] [CrossRef]
- Beaudry, M.S.; Wang, J.; Kieran, T.J.; Thomas, J.; Bayona-Vásquez, N.J.; Gao, B.; Devault, A.; Brunelle, B.; Lu, K.; Wang, J.-S.; et al. Improved Microbial Community Characterization of 16S rRNA via Metagenome Hybridization Capture Enrichment. Front. Microbiol. 2021, 12, 644662. [Google Scholar] [CrossRef]
- Guitor, A.K.; Raphenya, A.R.; Klunk, J.; Kuch, M.; Alcock, B.; Surette, M.G.; McArthur, A.G.; Poinar, H.N.; Wright, G.D. Capturing the Resistome: A Targeted Capture Method To Reveal Antibiotic Resistance Determinants in Metagenomes. Antimicrob. Agents Chemother. 2019, 64, e01324-19. [Google Scholar] [CrossRef]
- Andrew, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 January 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Pearman, W.S.; Freed, N.E.; Silander, O.K. Testing the advantages and disadvantages of short- and long- read eukaryotic metagenomics using simulated reads. BMC Bioinform. 2020, 21, 220. [Google Scholar] [CrossRef]
- Gehrig, J.L.; Portik, D.M.; Driscoll, M.D.; Jackson, E.; Chakraborty, S.; Gratalo, D.; Ashby, M.; Valladares, R. Finding the right fit: Evaluation of short-read and long-read sequencing approaches to maximize the utility of clinical microbiome data. Microb. Genom. 2022, 8, 000794. [Google Scholar] [CrossRef]
- Tedersoo, L.; Albertsen, M.; Anslan, S.; Callahan, B. Perspectives and Benefits of High-Throughput Long-Read Sequencing in Microbial Ecology. Appl. Environ. Microbiol. 2021, 87, e0062621. [Google Scholar] [CrossRef]
- Illumina, I. How Short Inserts Affect Sequencing Performance. Available online: https://support.illumina.com/bulletins/2020/12/how-short-inserts-affect-sequencing-performance.html (accessed on 30 August 2022).
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Minich, J.J.; Sanders, J.G.; Amir, A.; Humphrey, G.; Gilbert, J.A.; Knight, R. Quantifying and Understanding Well-to-Well Contamination in Microbiome Research. mSystems 2019, 4, e00186-19. [Google Scholar] [CrossRef]
- Salem, S.E.; Maddox, T.W.; Berg, A.; Antczak, P.; Ketley, J.M.; Williams, N.J.; Archer, D.C. Variation in faecal microbiota in a group of horses managed at pasture over a 12-month period. Sci. Rep. 2018, 8, 8510. [Google Scholar] [CrossRef]
- Johnson, A.; Biddle, A. 2 Evaluation of the microbiome effects of easy and hard keeper horses with the application of the Equine Keeper Status Scale. J. Equine Vet. Sci. 2021, 100, 103465. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Portillo, M.C.; Belda-Ferre, P.; Mira, A. Amplification by PCR artificially reduces the proportion of the rare biosphere in microbial communities. PLoS ONE 2012, 7, e29973. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.D.; Bloom, R.J.; Jiang, S.; Durand, H.K.; Dallow, E.; Mukherjee, S.; David, L.A. Measuring and mitigating PCR bias in microbiota datasets. PLoS Comput. Biol. 2021, 17, e1009113. [Google Scholar] [CrossRef]
- Sipos, R.; Székely, A.J.; Palatinszky, M.; Révész, S.; Márialigeti, K.; Nikolausz, M. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol. Ecol. 2007, 60, 341–350. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M.; Parfrey, L.W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 2018, 6, 41. [Google Scholar] [CrossRef]
- Felske, A.; Akkermans, A.D.; De Vos, W.M. Quantification of 16S rRNAs in complex bacterial communities by multiple competitive reverse transcription-PCR in temperature gradient gel electrophoresis fingerprints. Appl. Environ. Microbiol. 1998, 64, 4581–4587. [Google Scholar] [CrossRef]
- Morris, E.K.; Caruso, T.; Buscot, F.; Fischer, M.; Hancock, C.; Maier, T.S.; Meiners, T.; Müller, C.; Obermaier, E.; Prati, D.; et al. Choosing and using diversity indices: Insights for ecological applications from the German Biodiversity Exploratories. Ecol. Evol. 2014, 4, 3514–3524. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||||
|---|---|---|---|---|---|---|
| Genus level | ||||||
| TEHC | 16S Amplicon | |||||
| Sample | Shared | Exclusive | Total | Shared | Exclusive | Total |
| 1 | 135 (58%) | 99 (42%) | 234 | 135 (72%) | 53 (28%) | 188 |
| 2 | 143 (60%) | 94 (40%) | 237 | 143 (70%) | 60 (30%) | 203 |
| 3 | 152 (67%) | 76 (33%) | 228 | 152 (72%) | 58 (38%) | 210 |
| 4 | 150 (60%) | 99 (40%) | 249 | 150 (67%) | 75 (33%) | 225 |
| (B) | ||||||
| Species level | ||||||
| TEHC | 16S Amplicon | |||||
| Sample | Shared | Exclusive | Total | Shared | Exclusive | Total |
| 1 | 135 (72%) | 53 (28%) | 188 | 42 (71%) | 17 (29%) | 59 |
| 2 | 143 (70%) | 60 (30%) | 203 | 54 (73%) | 20 (27%) | 74 |
| 3 | 152 (72%) | 58 (38%) | 210 | 77 (81%) | 18 (19%) | 95 |
| 4 | 150 (67%) | 75 (33%) | 225 | 56 (70%) | 24 (30%) | 80 |
| Genus | 16S Amplicon | TEHC | Expected |
|---|---|---|---|
| Acinetobacter | 6.15 | 5.50 | 5 |
| Actinomyces | 0.63 | 5.08 | 5 |
| Bacillus | 0.04 | 0.18 | 5 |
| Bacteroides | 6.74 | 6.47 | 5 |
| Clostridium | 9.54 | 7.62 | 5 |
| Deinococcus | 3.27 | 3.72 | 5 |
| Enterococcus | 3.94 | 4.88 | 5 |
| Escherichia | 0.00 | 0.00 | 5 |
| Helicobacter | 12.19 | 5.98 | 5 |
| Lactobacillus | 1.81 | 5.25 | 5 |
| Listeria | 0.00 | 0.00 | 5 |
| Neisseria | 7.18 | 5.11 | 5 |
| Propionibacterium | 0.36 | 3.72 | 5 |
| Pseudomonas | 0.22 | 4.74 | 5 |
| Rhodobacter | 2.49 | 3.04 | 5 |
| Staphylococcus | 10.58 | 8.71 | 10 |
| Streptococcus | 21.77 | 14.89 | 15 |
| Prevotella | 1.01 | 0.01 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez Narváez, S.; Beaudry, M.S.; Norris, C.G.; Bartlett, P.B.; Glenn, T.C.; Sanchez, S. Improved Equine Fecal Microbiome Characterization Using Target Enrichment by Hybridization Capture. Animals 2024, 14, 445. https://doi.org/10.3390/ani14030445
Álvarez Narváez S, Beaudry MS, Norris CG, Bartlett PB, Glenn TC, Sanchez S. Improved Equine Fecal Microbiome Characterization Using Target Enrichment by Hybridization Capture. Animals. 2024; 14(3):445. https://doi.org/10.3390/ani14030445
Chicago/Turabian StyleÁlvarez Narváez, Sonsiray, Megan S. Beaudry, Connor G. Norris, Paula B. Bartlett, Travis C. Glenn, and Susan Sanchez. 2024. "Improved Equine Fecal Microbiome Characterization Using Target Enrichment by Hybridization Capture" Animals 14, no. 3: 445. https://doi.org/10.3390/ani14030445