
Characterization and Comparative Analysis of Whole-Transcriptome Sequencing in High- and Low-Fecundity Chongming White Goat Ovaries during the Estrus Phase
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Sample Preparation
2.3. miRNA Sequencing
2.4. circRNA Sequencing
2.5. mRNA and lncRNA Sequencing
2.6. Gene Co-Expression Analysis
2.7. Validation of RNA-Seq Results Analyzed Using Quantitative PCR (qPCR)
3. Results
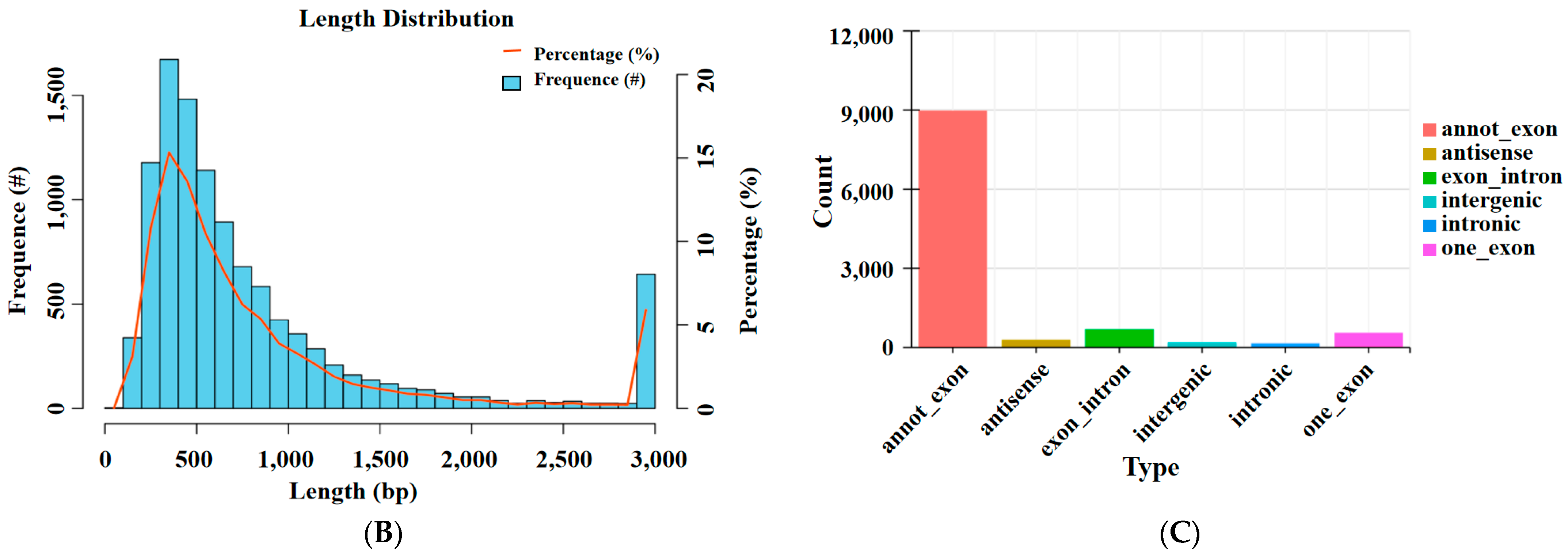
3.1. Overview of circRNA Sequencing
3.2. CircRNA Source Gene Analysis and Differentially Expressed DEcircRNAs Analysis
3.3. Sequencing and DEmiRNAs Analysis

3.4. Sequencing and DEmRNA and DElncRNA Analysis
3.5. CeRNA Regulatory Network
3.6. qPCR Validation of Differentially Expressed mRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Agossou, D.J.; Koluman, N. The effects of natural mating and artificial insemination using cryopreserved buck semen on reproductive performance in Alpine goats. Arch. Anim. Breed. 2018, 61, 459–461. [Google Scholar] [CrossRef]
- Esmaeili-Fard, S.M.; Gholizadeh, M.; Hafezian, S.H.; Abdollahi-Arpanahi, R. Genome-wide association study and pathway analysis identify NTRK2 as a novel candidate gene for litter size in sheep. PLoS ONE 2021, 16, e0244408. [Google Scholar] [CrossRef] [PubMed]
- Estefanía, G.; Mcguire, B.C.; Keating, A.F. Obesity alters the ovarian proteomic response to zearalenone exposure. Biol. Reprod. 2021, 1, 278–289. [Google Scholar]
- Kon, Y.; Islam, M.R.; Ichii, O.; Nakamura, T.; Hosny, Y. Developmental Changes of the Ovary in Neonatal Cotton Rat (Sigmodon hispidus). Front. Physiol. 2021, 11, 601927. [Google Scholar]
- Teena, R.; Dhamodharan, U.; Ali, D.; Rajesh, K.; Ramkumar, K.M. Gene Expression Profiling of Multiple Histone Deacetylases (HDAC) and Its Correlation with NRF2-Mediated Redox Regulation in the Pathogenesis of Diabetic Foot Ulcers. Biomolecules 2020, 10, 1466. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Bejugam, P.R.; Das, A.; Panda, A.C. Seeing Is Believing: Visualizing Circular RNAs. Non-Coding RNA 2020, 6, 45. [Google Scholar] [CrossRef]
- Wilusz, J.E. Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim. Biophys. 2015, 1859, 128–138. [Google Scholar] [CrossRef]
- Lu, T.; Zou, X.; Liu, G.; Deng, M.; Sun, B.; Guo, Y.; Liu, D.; Li, Y. A Preliminary Study on the Characteristics of microRNAs in Ovarian Stroma and Follicles of Chuanzhong Black Goat during Estrus. Genes 2020, 11, 970. [Google Scholar] [CrossRef]
- Zhao, Z.; Zou, X.; Lu, T.; Deng, M.; Liu, D. Identification of mRNAs and lncRNAs Involved in the Regulation of Follicle Development in Goat. Front. Genet. 2020, 11, 589076. [Google Scholar] [CrossRef]
- Tao, H.; Xiong, Q.; Zhang, F.; Zhang, N.; Liu, Y.; Suo, X.; Li, X.; Yang, Q.; Chen, M. Circular RNA profiling reveals chi_circ_0008219 function as microRNA sponges in pre-ovulatory ovarian follicles of goats (Capra hircus). Genomics 2018, 110, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, Z.; Guo, S.; Li, K.; Wang, P.; Fan, Y.; He, X.; Jiang, Y.; Lan, R.; Chen, S.; et al. Transcriptome analysis reveals key miRNA-mRNA pathways in ovarian tissues of yunshang black goats with different kidding numbers. Front. Endocrinol. 2022, 13, 883663. [Google Scholar] [CrossRef]
- Liang, C.; Han, M.C.; Zhou, Z.Y.; Liu, Y.F.; He, X.Y.; Jiang, Y.T.; Ouyang, Y.N.; Hong, Q.H.; Chu, M.X. Hypothalamic transcriptome analysis reveals the crucial microRNAs and mRNAs affecting litter size in goats. Front. Vet. Sci. 2021, 8, 74710010. [Google Scholar] [CrossRef] [PubMed]
- Kandoi, G.; Dickerson, J.A. Tissue-specific mouse mRNA isoform networks. Sci. Rep. 2019, 9, 13949. [Google Scholar] [CrossRef]
- Simoneau, J.; Dumontier, S.; Gosselin, R.; Scott, M.S. Current RNA-seq methodology reporting limits reproducibility. Brief. Bioinform. 2019, 22, 140–145. [Google Scholar] [CrossRef]
- Li, Q.; Pan, X.; Li, N.; Gong, W.; Yuan, X. Identification of circular RNAs in hypothalamus of gilts during the onset of puberty. Genes 2021, 12, 84. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Z.; He, X.; Tao, L.; Jiang, Y.; Lan, R.; Hong, Q.; Chu, M. Integrated analyses of miRNA-mRNA expression profiles of ovaries reveal the crucial interaction networks that regulate the prolificacy of goats in the follicular phase. BMC Genom. 2021, 22, 812. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Luo, Y.; Cui, L.; Chen, L.; Wang, L.; Ji, K.; Liu, H. Characterization of the myometrial transcriptome of long non-coding RNA genes in human labor by high-throughput RNA-seq. Reprod. Sci. 2022, 29, 2885–2893. [Google Scholar] [CrossRef]
- Wu, F.; Sun, H.; Lu, S.; Gou, X.; Pan, Y. Genetic diversity and selection signatures within diannan small-ear pigs revealed by next-generation sequencing. Front. Genet. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Li, T.S.; Li, X.N.; Chang, Z.J.; Fu, X.Y.; Liu, L. Identification and functional characterization of a novel interleukin 17 receptor: A possible mitogenic activation through ras/mitogen-activated protein kinase signaling pathway. Cell Signal 2006, 18, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Rauen, K.A.; Tidyman, W.E.; Estep, A.L.; Sampath, S.; Peltier, H.M.; Bale, S.J.; Lacassie, Y. Molecular and functional analysis of a novel MEK2 mutation in cardio-facio-cutaneous syndrome: Transmission through four generations. Am. J. Med. Genet. Part A 2010, 152A, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Laursen, L.; Inturi, R.; Stergaard, S.; Jemth, P. Determinants of affinity, specificity, and phase separation in a supramodule from Post-synaptic density protein 95. iScience 2022, 25, 105069. [Google Scholar] [CrossRef] [PubMed]
- Kiho, L.; Robert, P.; Campbell, R.E.; Bernhard, L.s.; Herbison, A.E. Knockdown of GABA(A) receptor signaling in GnRH neurons has minimal effects upon fertility. Endocrinology 2010, 151, 4428–4436. [Google Scholar]
- Yamagata, H.; Ogihara, H.; Matsuo, K.; Uchida, S.; Kobayashi, A.; Seki, T.; Kobayashi, M.; Harada, K.; Chen, C.; Miyata, S. Distinct epigenetic signatures between adult-onset and late-onset depression. Sci. Rep. 2021, 11, 2296. [Google Scholar] [CrossRef] [PubMed]
- Demars, M.P.; Morishita, H. Cortical parvalbumin and somatostatin GABA neurons express distinct endogenous modulators of nicotinic acetylcholine receptors. Mol. Brain 2014, 7, 75. [Google Scholar] [CrossRef]
- Mukhtar, T.; Breda, J.; Grison, A.; Karimaddini, Z.; Grobecker, P.; Iber, D.; Beisel, C.; Nimwegen, E.V.; Taylor, V. Tead transcription factors differentially regulate cortical development. Sci. Rep. 2020, 10, 4625. [Google Scholar] [CrossRef]
- Clark, K.L.; George, J.W.; Emilia, P.; Plewes, M.R.; Guohua, H.; Cheng, W.; Davis, J.S. Hippo signaling in the ovary: Emerging roles in development, fertility, and disease. Endocr. Rev. 2022, 43, 1074–1096. [Google Scholar] [CrossRef]
- Jaffe, L.A.; Egbert, J.R. Regulation of mammalian oocyte meiosis by intercellular communication within the ovarian follicle. Annu. Rev. Physiol. 2017, 79, 237–260. [Google Scholar] [CrossRef]
- Germano, G.; Porazzi, P.; Felix, C. Leukemia-associated transcription factor mllt3 is important for primitive erythroid development in zebrafish embryogenesis. Dev. Dyn. 2022, 251, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Biittner, N.; Johnsen, S.A.; Kuegler, S.; Vogel, T. Af9/Mllt3 interferes with Tbr1 expression through epigenetic modification of histone H3K79 during development of the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 7042–7047. [Google Scholar] [CrossRef]
- Giovanni, C.; Cristina, G.M.; Albertini, D.F.; Mariabeatrice, D.C.; Mario, M.R.; Elena, D.P.; Rubens, F. Contributions of the actin cytoskeleton to the emergence of polarity during maturation in human oocytes. Mol. Hum. Reprod. 2014, 20, 200–207. [Google Scholar]
- Coticchio, G.; Dal Canto, M.; Mignini Renzini, M.; Guglielmo, M.C.; Brambillasca, F.; Turchi, D.; Novara, P.V.; Fadini, R. Oocyte maturation: Gamete-somatic cells interactions, meiotic resumption, cytoskeletal dynamics and cytoplasmic reorganization. Hum. Reprod. Update 2015, 21, 427–454. [Google Scholar] [CrossRef]
- Sirotkin, A.V.; Ovcharenko, D.; Grossmann, R.; Lauková, M.; Mlyncek, M. Identification of MicroRNAs controlling human ovarian cell steroidogenesis via a genome-scale screen. J. Cell. Physiol. 2009, 219, 415–420. [Google Scholar] [CrossRef] [PubMed]
- An, X.P.; Hou, J.X.; Li, G.; Song, Y.X.; Wang, J.G.; Chen, Q.J.; Cui, Y.H.; Wang, Y.F.; Cao, B.Y. Polymorphism identification in the goat KITLG gene and association analysis with litter size. Anim. Genet. 2012, 43, 104–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Zhou, Z.N.; Tian, X.Z.; Yang, P.F.; Fu, K.B. CYP19A1 may influence lambing traits in goats by regulating the biological function of granulosa cells. Animals 2022, 12, 1911. [Google Scholar] [CrossRef]
- Tomar, A.; Uysal-Onganer, P.; Basnett, P.; Pati, U.; Roy, I. 3D disease modelling of hard and soft cancer using PHA-based scaffolds. Cancers 2022, 14, 3549. [Google Scholar] [CrossRef]
- Almiana, C.; Dubuisson, F.; Bauersachs, S.; Royer, E.; Mermillod, P.; Blesbois, E.; Guignot, F. Unveiling how vitrification affects the porcine blastocyst: Clues from a transcriptomic study. J. Anim. Sci. Biotechnol. 2022, 13, 46. [Google Scholar]
- Wang, Y.; Yang, H.M.; Cao, W.; Li, Y.B.; Wang, Z.Y. Deep sequencing identification of miRNAs in pigeon ovaries illuminated with monochromatic light. BMC Genom. 2018, 19, 446. [Google Scholar] [CrossRef]
- Tarko, A.; Štochmal’Ová, A.; Jedliková, K.; Hrabovszká, S.; Vachanová, A.; Harrath, A.H.; Alwasel, S.; Alrezaki, A.; Kotwica, J.; Balái, A. Effects of benzene, quercetin, and their combination on porcine ovarian cell proliferation, apoptosis, and hormone release. Arch. Anim. Breed. 2019, 62, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.P.; Zhang, Z.J.; Liu, Y.F.; Ren, C.H.; He, X.Y.; Jiang, Y.T.; Ouyang, Y.A.; Hong, Q.H.; Chu, M.X. Integrated analysis of mRNAs and long non-coding RNAs expression of oviduct that provides novel insights into the prolificacy mechanism of goat (Capra hircus). Genes 2022, 13, 1031. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Zacchi, L.F.; Schulz, B.L.; Moore, S.S.; Fortes, M.R.S. Adipose tissue proteomic analyses to study puberty in Brahman heifers. J. Anim. Sci. 2018, 96, 2392–2398. [Google Scholar] [CrossRef] [PubMed]
- Mcfee, R.M.; Artac, R.A.; Mcfee, R.M.; Clopton, D.T.; Smith, R.A.L.; Rozell, T.G.; Cupp, A.S. Inhibition of vascular endothelial growth factor receptor signal transduction blocks follicle progression but does not necessarily disrupt vascular development in perinatal rat ovaries. Biol. Reprod. 2009, 81, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Do, J.H.; Bae, S.; Bae, D.H.; Ahn, W.S. Identification of differentially expressed genes using an annealing control primer system in stage III serous ovarian carcinoma. BMC Cancer 2010, 10, 576. [Google Scholar] [CrossRef]
- Xu, Y.; Li, E.; Han, Y.; Chen, L.; Xie, Z. Differential expression of mRNAs encoding BMP/Smad pathway molecules in antral follicles of high- and low-fecundity Hu sheep. Anim. Reprod. Sci. 2010, 120, 47–55. [Google Scholar] [CrossRef]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Genome-wide analysis of miRNAs in the ovaries of Jining Grey and Laiwu Black goats to explore the regulation of fecundity. Sci. Rep. 2016, 6, 37983. [Google Scholar] [CrossRef]
- Lin, S.Y.; Morrison, J.R.; Phillips, D.J.; de Kretser, D.M. Regulation of ovarian function by the TGF-beta superfamily and follistatin. Reproduction 2003, 126, 133–148. [Google Scholar] [CrossRef]
- Duygu, S.H.; Ileana, C.; Castrillon, D.H.; Joan, J. Control of oocyte reawakening by Kit. PLoS Genet. 2016, 12, e1006215. [Google Scholar]
- Li, X.; Zhong, Y.; Zhou, W.; Song, Y.; Li, W.; Jin, Q.; Gao, T.; Zhang, L.; Xie, M. Low-intensity pulsed ultrasound (LIPUS) enhances the anti-inflammatory effects of bone marrow mesenchymal stem cells (BMSCs)-derived extracellular vesicles. Cell. Mol. Biol. Lett. 2023, 28, 9. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.H.; Yang, C.Y.; Yeh, T.H.; Huang, Y.C.; Wang, T.W.; Yu, J.Y. The Hippo pathway acts downstream of the Hedgehog signaling to regulate follicle stem cell maintenance in the Drosophila ovary. Sci. Rep. 2017, 7, 4480. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | GenBank Accession No. | Primer Sequences (5-3) | Tm (°C) | Length (bp) |
|---|---|---|---|---|
| SCARB1 | XM_018061401.1 | F: GGCACAAGAGCAACATCACC | 59 | 145 |
| R: ATCGACGCACTCAAGACCAG | ||||
| CYP19A1 | NM_001285747.1 | F: GCATCTGGACAGGTTGGAGG | 61 | 344 |
| R: CAAAAATCAACTCAGTGGCGAA | ||||
| LDLR | XM_005682375.3 | F: CGCCTACCTCTTCTTCACCAA | 60 | 105 |
| R: TCCAGGGCAACCACATTCTT | ||||
| SDC4 | XM_018057920.1 | F: TGCACCCCTTGGTCCCTAT | 60 | 203 |
| R: TGCCAGCACCTCTGTCCTCT | ||||
| LOC102181202 | XM_018038833.1 | F: CTGTGATCTGGAGGAAGAAGC | 61 | 78 |
| R: CCTGGGCACTGTCATTGTT | ||||
| DHCR24 | XM_005678362.3 | F: TGAAGACAAACCGAGAGGGC | 60 | 133 |
| R: CCAAAGAGGTAGCGGAAGATG | ||||
| TNXB | XM_018038915.1 | F: ATACACAGACGAGGATGGGCA | 60 | 217 |
| R: CGGGGTTTGGGTGGTAGAGA | ||||
| SKP1 | XM_018050387.1 | F: TTGGAAGATTTGGGAATGGAT | 59 | 208 |
| R: GTGTCCCTTGGTCAACTTTCAG | ||||
| VEGFA | XM_018038496.1 | F: GGGGCTGCTGTAATGACGA | 60 | 97 |
| R: TGCTGGCTTTGGTGAGGTTT | ||||
| TIMP1 | XM_005700836.3 | F: CACAGGTCCCAGAATCGCA | 60 | 159 |
| R: TTCCTCACAACCAGCAGCATA | ||||
| SOCS3 | XM_018063683.1 | F: CGAGAAGATCCCTCTGGTGTT | 59 | 107 |
| R: CTTTCTCGTAGGAGTCCAGGTG | ||||
| ACSL4 | XM_018044177.1 | F: AGTCCATATCGTTCTGTCACGC | 61 | 123 |
| R: TCCCAGGCTCTCCTTCTTCC | ||||
| SLC2A1 | NM_001314223.1 | F: CGGCAGATGATGCGAGAGA | 62 | 229 |
| R: GCGACACGACAGTGAAGGCT | ||||
| RCAN1 | XM_018055819.1 | F: GCTTCAAACGGGTCAGAATAAA | 59 | 140 |
| R: CAGGTGCGAACTGCCTATGT | ||||
| RPL19 | XM_005693740.3 | F: ATCGCCAATGCCAACTC | 60 | 154 |
| R: CCTTTCGCTTACCTATACC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.; Sun, L.; Dai, J.; Lv, Y.; Liao, R.; Shen, X.; Gao, J. Characterization and Comparative Analysis of Whole-Transcriptome Sequencing in High- and Low-Fecundity Chongming White Goat Ovaries during the Estrus Phase. Animals 2024, 14, 988. https://doi.org/10.3390/ani14070988
Lin Y, Sun L, Dai J, Lv Y, Liao R, Shen X, Gao J. Characterization and Comparative Analysis of Whole-Transcriptome Sequencing in High- and Low-Fecundity Chongming White Goat Ovaries during the Estrus Phase. Animals. 2024; 14(7):988. https://doi.org/10.3390/ani14070988
Chicago/Turabian StyleLin, Yuexia, Lingwei Sun, Jianjun Dai, Yuhua Lv, Rongrong Liao, Xiaohui Shen, and Jun Gao. 2024. "Characterization and Comparative Analysis of Whole-Transcriptome Sequencing in High- and Low-Fecundity Chongming White Goat Ovaries during the Estrus Phase" Animals 14, no. 7: 988. https://doi.org/10.3390/ani14070988
APA StyleLin, Y., Sun, L., Dai, J., Lv, Y., Liao, R., Shen, X., & Gao, J. (2024). Characterization and Comparative Analysis of Whole-Transcriptome Sequencing in High- and Low-Fecundity Chongming White Goat Ovaries during the Estrus Phase. Animals, 14(7), 988. https://doi.org/10.3390/ani14070988