Simple Summary
This study utilized data from 936 pigs for genome-wide association studies (GWAS) of four semen traits (sperm motility, sperm progressive motility, sperm abnormality rate, and total sperm count), as well as data from 5457 pigs from FarmGTEx for transcriptome analysis. The results revealed 16, 9, and 12 significant single nucleotide polymorphisms (SNPs) associated with semen traits in Duroc, Landrace, and Yorkshire pigs, with 7, 5, and 7 candidate genes identified in these breeds, potentially linked to mammalian spermatogenesis, testicular function, and male fertility. This research deepened the understanding of semen trait genetics and provided insights for enhancing semen quality in these pig breeds.
Abstract
High-quality semen is an essential factor for the success of artificial insemination, and revealing the genetic structure of pig semen traits helps improve semen quality. This study aimed to identify candidate genes associated with semen traits in three pig breeds (Duroc, Landrace, and Yorkshire) through weighted GWAS and multi-tissue transcriptome analysis. In this study, to identify candidate genes associated with semen traits in Duroc, Landrace, and Yorkshire, we performed weighted GWAS in four traits (sperm motility, sperm progressive motility, sperm abnormality rate, and total sperm count) using 936 pigs and multi-tissue transcriptome analysis using 34 tissues RNA-seq data of 5457 pigs from FarmGTEx. It was found that 16, 9, and 12 significant SNPs associated with semen traits were identified in Duroc, Landrace, and Yorkshire, with corresponding 7, 5, and 7 candidate genes in these three breeds, respectively, which may be involved in mammal spermatogenesis, testicular function, and male fertility. Moreover, we not only found the same candidate gene DNAI2 as in previous studies but also found two new candidate genes PNLDC1 and RSPH3, which were identified simultaneously in both Landrace and Yorkshire. By integrating the GWAS and multi-tissue transcriptome analysis results, we found that candidate genes associated with semen traits of three pig breeds were highly expressed in the testis tissue. The three genotypes of rs320928244 had significant effects on the expression of the DYNLT1 gene in the testis tissue of Landrace. These results together showed that these candidate genes were mainly related to sperm motility defects. This study helps deepen the understanding of the genetic basis of semen traits and provides a theoretical foundation for improving the semen quality of Duroc, Landrace, and Yorkshire breeds.
1. Introduction
With the rapid development of artificial insemination (AI), it has become important to select boars with outstanding semen quality to improve fertilization outcomes [1]. Traditional breeding schemes have focused on selecting pigs with excellent growth performance, carcass performance, and female reproductive traits while neglecting the potential benefits of semen traits on reproductive performance [2]. In recent years, with the rapid development of high-throughput genotyping and molecular techniques, genetic markers have gradually been utilized in genetic evaluation. There has been increasing interest in studying the molecular processes and genetic mechanisms that affect semen traits [3]. By revealing the genetic structure and candidate genes associated with pig semen traits, the efficiency of genome selection for target traits can be improved.
Genome-wide association studies (GWASs) commonly use high-throughput genotyping technology to assay single nucleotide polymorphisms (SNPs) and associate them with traits of interest [4]. At present, several studies have reported candidate genes and markers associated with pig semen traits by GWAS. Diniz et al. [5] reported the MTFMT gene associated with sperm motility traits in Landrace. Marques et al. [6] reported six/five candidate genes associated with semen traits in Landrace/Yorkshire. Godia et al. [7] identify candidate genes associated with semen traits and a series of mRNAs linked to sperm biology in Pietrain pigs by genome and transcriptome data. Additionally, Gao et al. [8], Zhao et al. [9], Mei et al. [10], and Zhang et al. [2] reported some candidate genes associated with semen traits in Duroc.
Previous GWAS studies in pig semen traits were primarily performed using traditional single SNPs GWAS and weighted single-step GWAS. Weighted single-step GWAS illustrates genetic variation through windows instead of single SNPs [6,11]. Because of the importance of semen quality in pig breeding, understanding the significant effects of single SNPs is crucial to designing selection programs. The weighted GWAS, proposed by Li et al. [12], is a method that assigns weights to residual variances, aiming to reduce stratification and stabilize solutions in GWAS. The weighted GWAS may outperform traditional GWAS methods when the number of animals with both phenotypes and genotypes is small. This method has been successfully applied to milk production traits [12]. In GWAS, the dependent variable must be a single value, whereas de-regressed estimated breeding values (DEBVs) combine repeated measures into a single value. DEBVs have proven to be effective response variables in GWAS studies of cattle milk production [13] and pig semen traits [10].
The integration of GWAS and multi-tissue transcriptome analysis helps understand the genetic structure of complex traits in cattle [14] and pigs [15]. To date, few studies have used genetics and transcriptome information to study the genetic background of pig semen traits. Godia et al. [7] identified three trans-expression QTL involving the candidate genes through the integration of GWAS (288 pigs) and sperm RNA sequencing (RNA-seq) data (35 pigs) analysis. The integration of GWAS and multi-tissue transcriptome analysis can provide a more intuitive understanding of the expression and biological significance of candidate genes in pig tissues.
In this study, we performed weighted GWAS using 936 pigs and multi-tissue transcriptome analysis using 34 tissues with 5457 RNA-seq data from FarmGTEx (http://piggtex.farmgtex.org/, accessed on 8 April 2024). The aim was to identify genetic variants and candidate genes associated with semen traits in three pig breeds. Subsequently, we analyzed the gene expression levels of candidate genes that could have the most significant impact on spermatogenesis and male fertility in 34 pig tissues. To gain a deeper understanding of the genetic mechanisms underlying pig semen traits, we performed a post-GWAS analysis that involved Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG). This analysis helps us to understand the biological processes and functional annotations of the candidate genes.
2. Material and Methods
2.1. Population and Phenotype Data
From August 2022 to February 2024, a total of 28,321 semen records were collected from 936 boars (382 Duroc, 290 Landrace, and 264 Yorkshire) at one artificial insemination station (Guangdong Guyue Technology Co., Ltd., Guangzhou, China). The boar populations were reared in individual fences with ad libitum access to drinking water, 14 h of daily light, and a daily diet of 2.5 kg, with adjustments made for individual boars based on their actual feed intake. Semen was collected every morning. Boars were introduced to the artificial insemination station from the same breeding farm at 4 months of age. These individuals shared similar genetic backgrounds, ages, and rearing conditions, which ensured consistency in evaluating semen traits and minimized external influences such as age differences or environmental stress. The pedigree file included 961 individual animals and four-generation pedigree records. The age of ejaculation for the boars ranged from 6.47 to 23.27 months. Four semen traits were measured, including sperm motility (SPMOT), sperm progressive motility (SPPMOT), sperm abnormality rate (SPABR), and total sperm count (SPCOUNT). After the semen was collected, it was immediately stored in an insulated bucket and kept at a temperature of 37 ± 1 °C, using a water bath in the laboratory. Subsequently, the SPMOT, SPPMOT, and SPABR of the original semen were measured using the Spain MagaporGesipor3.0 CASA system (CASA system analyzes SPMOT by tracking the movement of individual sperm cells in the sample, which refers to the proportion of motile sperm cells to the total number of sperm. SPPMOT: the proportion of sperm cells that move in a straight line. SPABR: the proportion of sperm cells with abnormal morphology. SPCOUNT: the total number of sperm cells in semen). The process of measuring semen traits typically takes approximately 10 min from the collection of the original semen. The SPCOUNT was calculated by multiplying the semen volume (mL) by the semen concentration (106/mL). The phenotypic values of semen traits were multiply compared using the “agricolae” package of R (v4.0.3) software.
The summary statistics of pig semen traits for three pig breeds are shown in Table 1. Referring to the research of Marques et al. [16] and Wang et al. [17], the quality control of phenotypes was as follows: (1) semen records with semen collection times less than 5 were excluded; (2) semen records with semen volume ≤ 50 mL were removed; (3) semen records with sperm motility < 10% were removed; (4) semen records with adjacent semen collection interval > 60 days or equal to 0 days were removed.

Table 1.
Descriptive statistics and heritabilities of the semen traits for three pig breeds.
2.2. Genotype Data
Genomic DNA was extracted and purified from semen samples (The semen samples were centrifuged at low speed to collect sperm) of 850 pigs, which included 361 Duroc, 257 Landrace, and 232 Yorkshire, using the TIANcombi DNA Lyse&Det PCR Kit (TIANGEN Biotech Co., Ltd. in Beijing, China). These pigs were genotyped using the 50 k SNP array (KPS Porcine Breeding Chip v1, Beijing, China), which contained 57,466 SNPs. We removed 12,213 SNPs that were not mapped to the reference genome (Sus scrofa 11.1). To detect outliers, we performed principal components (PCs) analysis on the genotype data using the R package Sommer [18]. For each breed, we excluded SNPs with a call rate lower than 0.9, a minor allele frequency (MAF) lower than 0.01, and a Hardy–Weinberg equilibrium (HWE) lower than 1 × 10−6 using PLINK v1.90 [19]. Finally, we retained 31,618 SNPs for Duroc (361 pigs), 33,704 SNPs for Landrace (257 pigs), and 39,925 SNPs for Yorkshire (232 pigs), respectively.
2.3. Variance Component Estimation and DEBV Calculation
The variance components of semen traits for each breed were computed using the average information restricted maximum likelihood (AI-REML) by DMU (version 6-R5-2-EM64T) software [20]. The mixed linear model was as follows:
where is the vector of phenotypes; is the vector of phenotype means; is the vector of fixed effects (overall mean year-season of ejaculation and birth parity of boar, these fixed effects had significant effects on semen quality of boars by F-test); N(0,) is the vector of additive genetic effects, is the additive genetic variance, is the pedigree matrices; N(0,I ) is the vector of permanent environment effects; is the variance of permanent environmental effects; , and are the design matrix corresponding to , , and , respectively. The covariates and represent the months of age and the intervals between ejaculations, respectively. N(0, I) is the residual effect vector, is the residual variance, is the identity matrix.
DEBVs were calculated according to VanRaden [21]. The formulas are described as follows:
where is the daughter equivalents; is the reliability of ; is the heritability of semen traits; , , , and PA are individual animal, sire, dam, and parent average, respectively.
2.4. Weighted Genome-Wide Association Study
Single trait-weighted GWAS was carried out by a mixed-model approach using MMAP (mmap.2021_08_19_22_30.intel, 2021) [22] software. The mixed model is as follows:
where is the vector of DEBVs for each semen trait; X is a matrix of genotypes (with 0, 1, or 2 represented genotypes AA, Aa, and aa, respectively), and is a vector of marker effects; g~N(0, ) is the vector of polygenic effects accounting for population structure, where is the genomic relationship matrix built by using all markers and is the genetic variance; and is the vector for residual effect, where is residual variances, R is a diagonal matrix weighted by the reliability of DEBVs and heritability.
Li et al. [12] demonstrated that considering the residual variances in the weighting process can effectively minimize stratification and enhance the stability of the solution. The weights utilized in R were associated with the reliability of conventional DEBVs () and the reliabilities of traditional PA [12]:
where is the heritability for each trait; is the proportion of genetic variation that cannot be explained by SNPs, when = 0.1, the DEBVs have high reliability; was computed as a function of daughter equivalents (DE):
and daughter equivalents for DEBV and PA were calculated as:
The genomic inflation factor (λ) was calculated by R software [23] from the raw p-values of the GWAS results [24]. The genome-wide false discovery rate (FDR) was applied to avoid false positives caused by multiple tests. The FDR was computed using the R package qvalue (v2.30.0, https://github.com/StoreyLab/qvalue, accessed on 16 March 2024). The threshold for significant SNPs was defined as FDR < 0.05. Manhattan plots and Quantile–Quantile (QQ) plots were performed using the R program. The network analysis and pathway diagrams of candidate genes were drawn using the R package clusterProfiler [25].
2.5. Gene Annotation and Functional Enrichment Analysis
The GWAS regions were identified based on the Sus scrofa 11.1 (release 106) database available at Ensembl (https://asia.ensembl.org/Sus_scrofa/Info/Index, accessed on 25 March 2024) by screening a distance of 1 Mb around the significant SNPs, referring to Hering et al. [26]. The function of candidate genes within the GWAS regions was manually identified at the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov, accessed on 2 April 2024). The Database for Annotation, Visualization, and Integrated Discovery (DAVID, https://david.ncifcrf.gov/tools.jsp, accessed on 6 April 2024) was used to perform the GO and KEGG enrichment analysis of candidate genes.
2.6. Integrative of GWAS Results and RNA-Seq Data
Genome and RNA-seq data from 34 tissues (5457 pigs) were downloaded from the Pig Genotype-Tissue Expression (http://piggtex.farmgtex.org/, accessed on 8 April 2024). In short, RNA-seq data were used to calculate gene expression levels (Transcripts per million, TPM) in 34 tissues. GWAS was then performed using gene expression levels as response variables and genomic data to identify expression quantitative trait locus (eQTL). By integrating the GWAS and eQTL results, we further investigated whether the significant SNPs affected gene expression in specific tissues. We recorded the genotype data (the 0, 1, and 2 represented genotypes AA, Aa, and aa, respectively) and then performed multiple comparisons using the least significant difference method based on the three genotypes of the significant SNPs and the expression levels of candidate genes. The LD between the significant SNPs of GWAS results and the significant eQTL of RNA-seq results was assessed using PLINK v1.9 [19] with the default parameters (“--ld-snp” and “--ld-window-kb”). The code for calculating TPM is available at the FarmGTEx Github website (https://github.com/FarmGTEx/PigGTEx-Pipeline-v0, accessed on 8 April 2024).
3. Results
3.1. Phenotypic Distribution and Variance Components Estimation
The average semen collection times for Duroc, Landrace, and Yorkshire were 37.12 ± 13.38, 27.54 ± 16.02, and 23.31 ± 16.48 times, respectively (Figure S1). The descriptive statistics and heritabilities of four semen traits are shown in Table 1. These findings suggest that the semen traits of Duroc, Landrace, and Yorkshire are moderately heritabilities, with estimations ranging from 0.10 to 0.35. Notably, the heritabilities of semen traits in Duroc were generally higher than those in Landrace and Yorkshire, except for SPPMOT. Table S1 provides more detailed information on the estimation of variance components for semen traits in each breed, including additive variance, permanent environmental effect variance, and residual variance. The distributions of DEBVs for semen traits in the three breeds are shown in Table S2 and Figure S2, indicating a normal distribution.
3.2. Weight Genome-Wide Association Study and Mining Candidate Genes
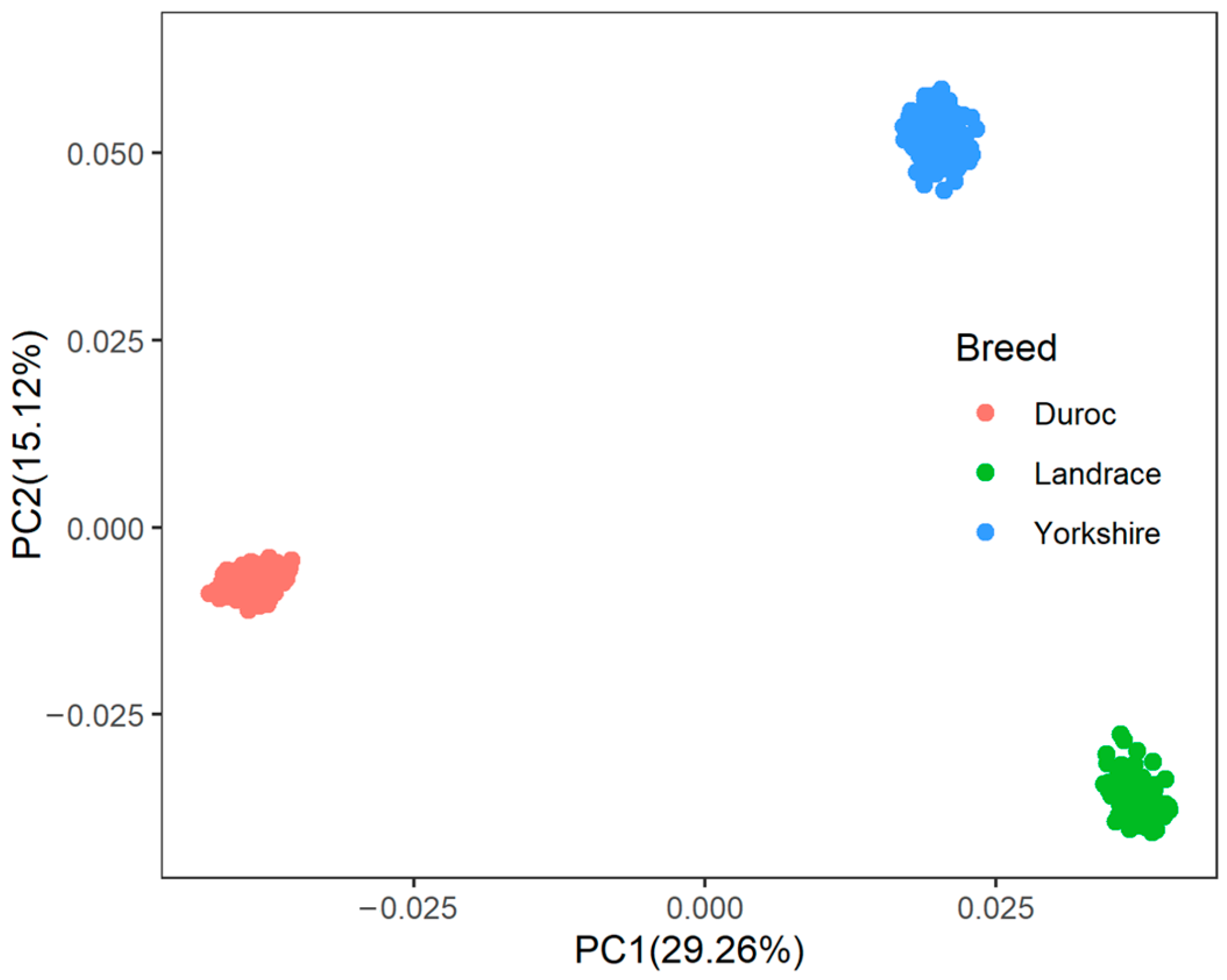
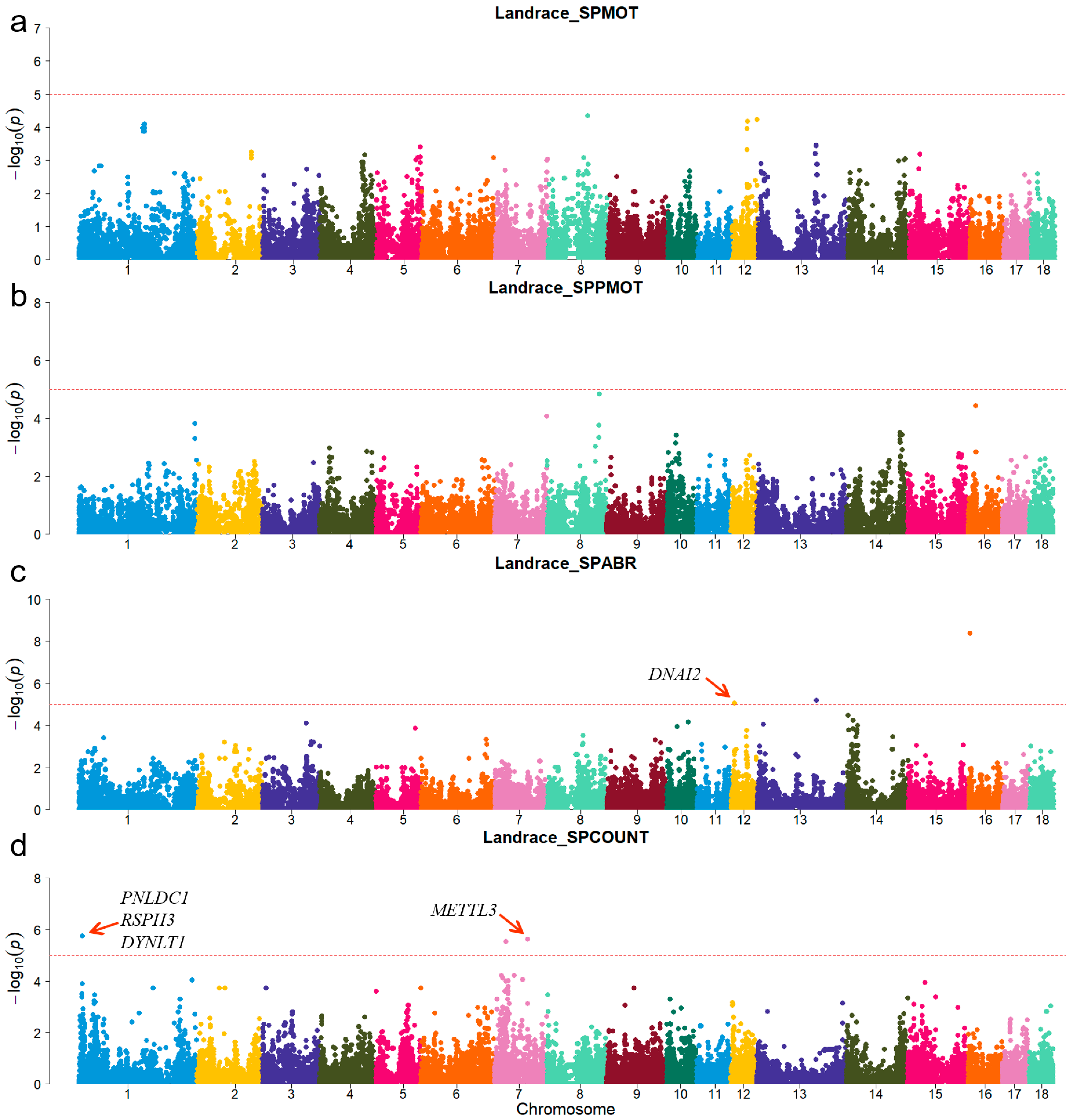
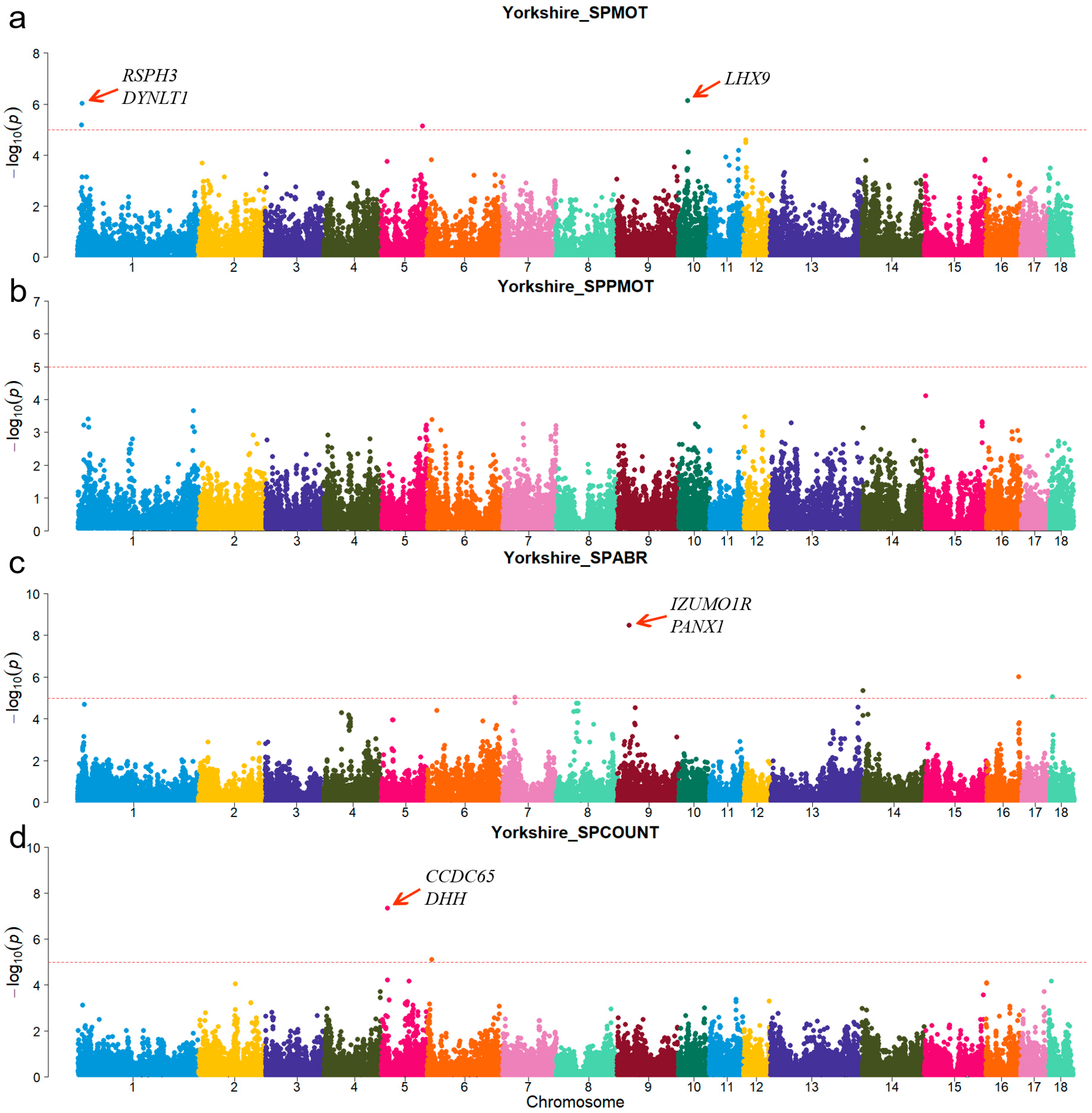
A principal component analysis was performed based on the genotypes of each breed (Figure 1). The first two genotype principal components (PCs) explained 29.26% and 15.12% of the total variance, respectively. Table S3 shows all significant SNPs and candidate genes associated with semen traits in the three pig breeds. Specifically, a total of 16, 9, and 12 significant SNPs, as well as 136, 97, and 152 candidate genes, were associated with semen traits in Duroc, Landrace, and Yorkshire, respectively. Table 2 shows promising candidate genes associated with semen traits in three pig breeds, which may be involved in spermiogenesis, and testes functioning. The table also provides details of the significant SNPs. The MAF distributions of the significant SNPs ranged from 0.15 to 0.36. The genomic inflation factors (λ) for four semen traits were close to 1, ranging from 0.89 to 1.09 (Figure S3). The candidate genes RSPH3 and DYNLT1, which were associated with the SPCOUNT trait in Landrace and the SPMOT trait in Yorkshire, were identified in the same region (8.24~8.56 Mb on chromosome 1).
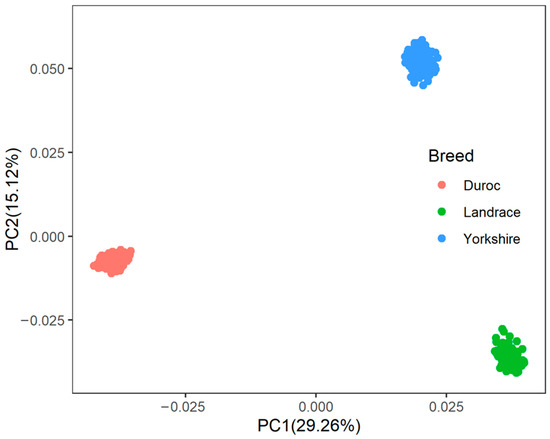
Figure 1.
The population structure of three pig breeds. PC1 = the first principal component, PC2 = the second principal component.
Table 2.
The significant SNPs and candidate genes of semen traits in three pig breeds.
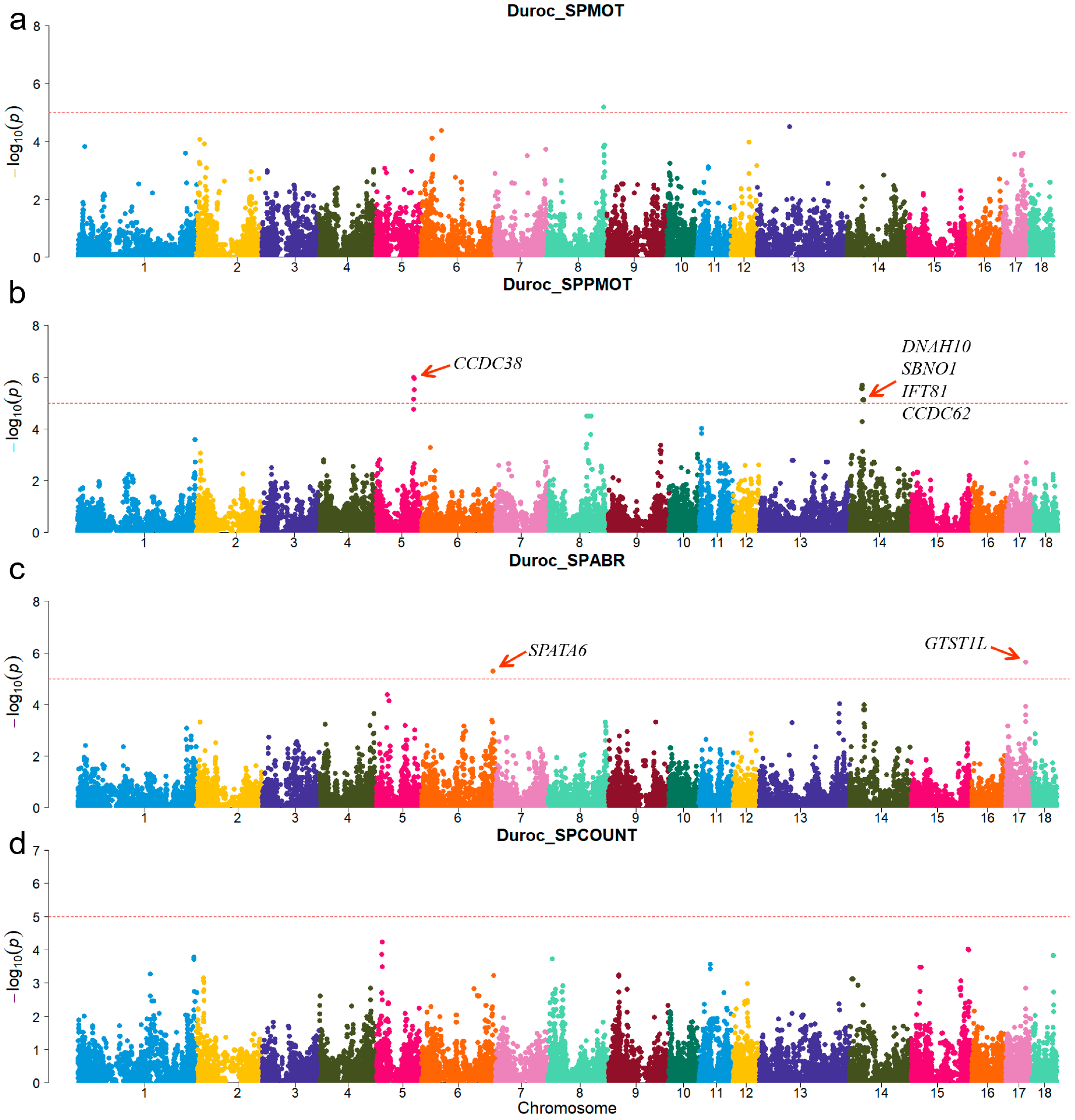
For Duroc, candidate genes coiled-coil domain containing 38 (CCDC38), Dynein axonemal heavy chain 10 (DNAH10), strawberry notch homolog 1 (SBNO1), coiled-coil domain containing 62 (CCDC62), and intraflagellar transport 81 (IFT81) were identified for the SPPMOT trait, the candidate genes spermatogenesis associated 6 (SPATA6) and gametocyte-specific factor 1 like (GTSF1L) were identified for the SPABR trait (Table 2 and Figure 2). For Landrace, candidate gene dynein axonemal intermediate chain 2 (DNAI2) was identified for the SPABR trait, the candidate genes methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit (METTL3), PARN-like ribonuclease domain containing exonuclease 1 (PNLDC1), radial spoke head 3 (RSPH3), and dynein light chain Tctex-type 1 (DYNLT1) were identified for the SPCOUNT trait (Table 2 and Figure 3). For Yorkshire, candidate genes RSPH3, DYNLT1, and LIM homeobox 9 (LHX9) were identified for the SPMOT trait, the candidate genes IZUMO1 receptor, JUNO (IZUMO1R) and pannexin 1 (PANX1) were identified for the SPABR trait, the candidate genes desert hedgehog signaling molecule (DHH) and coiled-coil domain containing 65 (CCDC65) were identified for the SPCOUNT trait (Table 2 and Figure 4). These candidate genes have shown promising associated with pig semen traits according to the NCBI annotation.
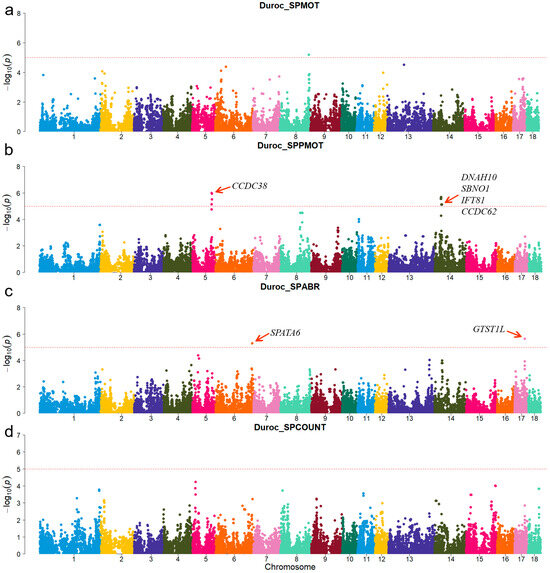
Figure 2.
Manhattan plots of semen-trait GWAS results in Duroc pigs. (a–d) are the SPMOT, SPPMOT, SPABR, and SPCOUNT, respectively.
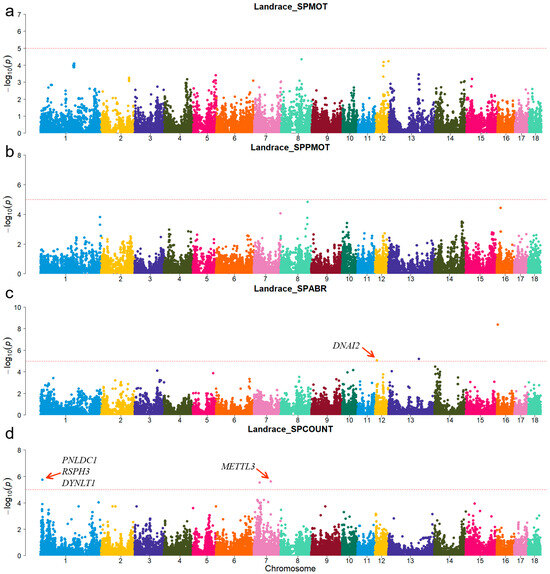
Figure 3.
Manhattan plots of semen-trait GWAS results in Landrace pigs. (a–d) are the SPMOT, SPPMOT, SPABR, and SPCOUNT, respectively.
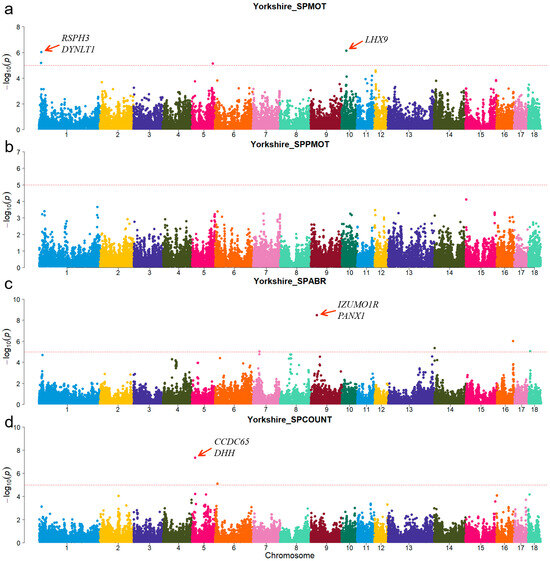
Figure 4.
Manhattan plots of semen-trait GWAS results in Yorkshire pigs. (a–d) are the SPMOT, SPPMOT, SPABR, and SPCOUNT, respectively.
3.3. Functional Enrichment Analyses of Candidate Genes
Table 3 shows the enriched GO terms and KEGG pathways of all candidate genes associated with semen traits in three pig breeds. These biological pathways include centrosome (GO:0005813), centriole (GO:0005814), smoothened signaling pathway (GO:0007224), axoneme (GO:0005930), peptidase activity (GO:0008233), ciliary basal body (GO:0036064), cilium assembly (GO:0060271), cilium (GO:0005929), fusion of sperm to egg plasma membrane (GO:0007342), and amyotrophic lateral sclerosis (ssc05014). The candidate genes DNAH10 (Duroc), DNAI2 (Landrace), and CCDC65 (Yorkshire) are involved in the biological process of the axoneme. CCDC38 and IFT81 (Duroc) are involved in the biological process of centrosomes. IFT81 (Duroc) and CCDC65 (Yorkshire) are involved in the biological process of the ciliary basal body. DNAH10 (Duroc) and DNAI2 (Landrace) are involved in the biological process of amyotrophic lateral sclerosis.
Table 3.
GO terms and KEGG pathways where the candidate genes were significantly (p < 0.05) enriched.
3.4. GWAS and Transcriptome Co-Localization Analysis
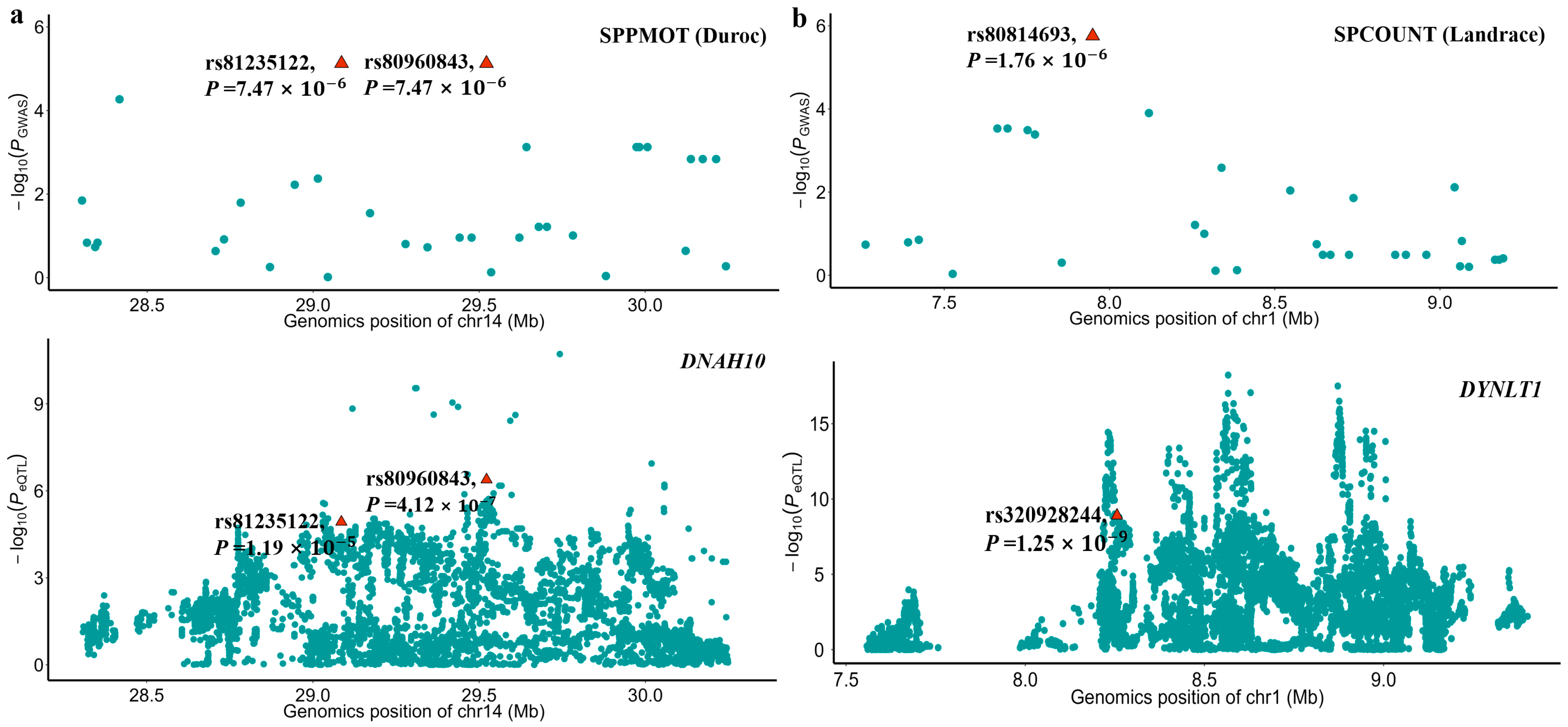
The sample sizes of RNA-seq data for each tissue were shown in Figure S4 and ranged from 43 to 1281. By integrating the GWAS and the eQTL results, we found that the significant SNPs, rs80960843 and rs81235122 of SPPMOT, were also identified as eQTL of the DNAH10 gene in testis tissue. This indicates that the two significant SNPs might affect the phenotype by regulating the expression of the DNAH10 gene. Additionally, the SNP rs320928244, which has a high LD (R2 = 0.929) with the significant SNP rs80814693 of SPCOUNT in Landrace, played a significant role in regulating the expression of the DYNLT1 gene in testis tissue (Figure 5).
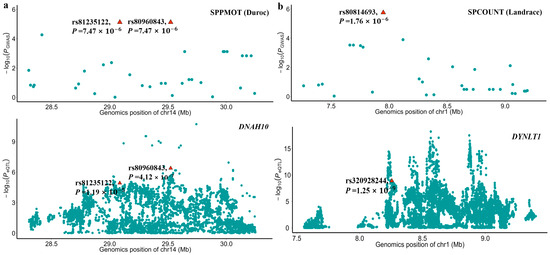
Figure 5.
Local Manhattan plots of GWAS and eQTL results. (a) The top local Manhattan plot depicted the GWAS of SPPMOT, highlighting the significant SNPs rs81235122 and rs80960843. The bottom local Manhattan plot shows the eQTL mapping of DNAH10 for the pig testis tissue. (b) The top local Manhattan plot depicted the GWAS of SPCOUNT, highlighting the significant SNPs rs80814693. The bottom local Manhattan plot shows that the SNP rs320928244 was the eQTL mapping of DYNLT1 in pig testis tissue. The SNP rs320928244 has a high LD (R2 = 0.929) with the significant SNP rs80814693. The −log10 p-values are shown on the y-axis.
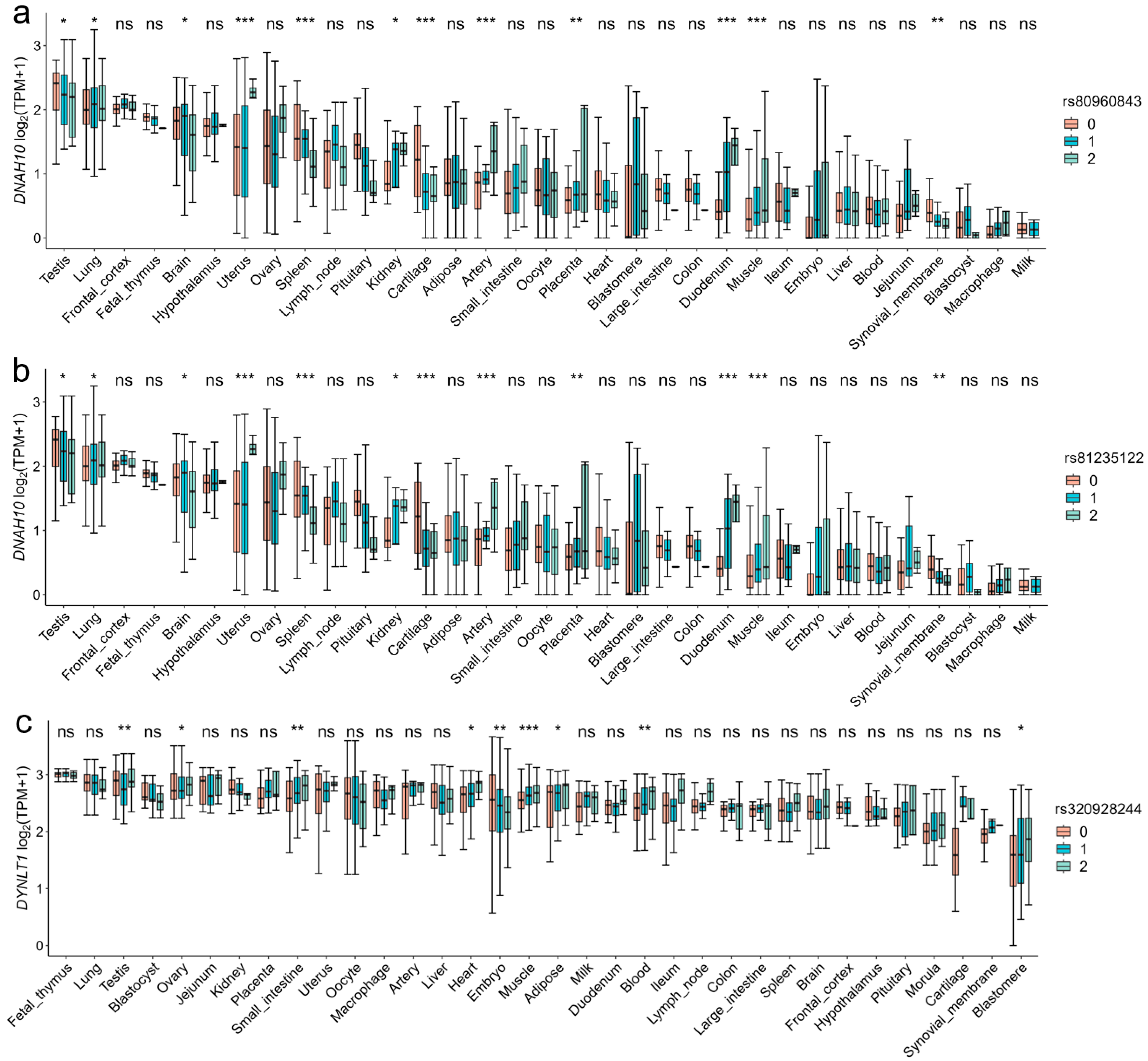
By calculating the TPM gene expression levels of candidate genes in 34 tissues, we found that the candidate genes CCDC38, DNAH10, SBNO1, CCDC62, IFT81, SPATA6 and GTSF1L in Duroc, DNAI2, METTL3, PNLDC1, RSPH3, and DYNLT1 in Landrace, and RSPH3, DYNLT1, LHX9, PANX1, DHH, and CCDC65 in Yorkshire exhibited highly expression levels in testis tissue (Figure S5). The three genotypes of the SNPs rs81235122, rs80960843, and rs320928244 were also found to exhibit high expression levels in pig testis tissue. Furthermore, we found significant differences in the expression of the DYNLT1 gene among the three genotypes of the SNP rs320928244 in pig testis tissue (Figure 6).
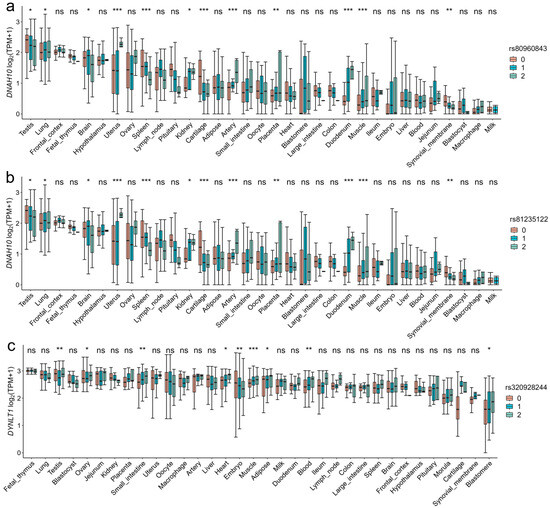
Figure 6.
Effects of three genotypes of significant SNPs on gene expression levels in pig-specific tissues. (a,b) The distribution of the DNAH10 gene expression for the significant SNPs (rs80960843 and rs81235122) across 34 tissues. (c) The distribution of the DYNLT1 gene expression for the SNP rs320928244 across 34 tissues. The SNP rs320928244 has a high LD (R2 = 0.929) with the significant SNP rs80814693 of DYNLT1. The y-axis represents the gene expression levels, while the x-axis represents different tissues. Significant differences between genotypes (0, 1, and 2 represented genotypes AA, Aa, and aa, respectively) and genotype expression are obtained using the least significant difference. ns means p > 0.05, * means p < 0.05, ** means p < 0.01, *** means p < 0.001.
4. Discussion
In this study, we performed a weighted genome-wide association study for the semen traits and multi-tissue transcriptome analysis in three pig breeds. Among these, 5457 RNA-seq data containing multiple pig breeds were obtained from FarmGTEx databases. The PigGTEx Phase 1 (Pilot) relied on publicly available RNA-Seq and WGS datasets to establish a foundational resource for tissue-specific gene expression in pigs. Despite its significance, this phase was constrained by several data gaps, such as incomplete coverage across tissues and breeds, and variability in data quality among sources. To address these challenges, multiple strategies were employed. Normalization techniques were applied to RNA-Seq data to reduce inter-study variability, while missing genomic information was inferred through imputation methods based on high-quality reference panels. Where possible, supplementary datasets and insights from related species were incorporated to provide a more holistic perspective. These efforts were particularly impactful for tissues of the hypothalamus–pituitary–gonadal axis, where data reliability allowed for more detailed analyses of gene expression patterns critical to reproductive traits. However, the analysis was limited for tissues and breeds with lower data quality or availability, and these gaps were noted as areas for improvement in future phases. While these strategies helped mitigate biases, the limitations underscore the need for continued data collection and integration in subsequent PigGTEx initiatives to achieve a truly comprehensive tissue-specific atlas for pigs.
The variance components results indicated that semen traits were moderate heritabilities traits, similar to the finding of Marques et al. [6], and higher than those reported by Gao et al. [8]. Moderate heritability indicates that semen traits have large genetic variation and can be selected. Additionally, we found that the heritabilities of semen traits in Duroc were higher than those in Landrace and Yorkshire, except for SPPMOT, which was consistent with the findings of Li et al. [27]. Therefore, the selection strategies of semen traits in three breeds should be considered separately. Furthermore, the results of principal components analysis indicated the absence of outliers, possibly due to the introduction of boar populations from the same pasture, which possesses a homogenous genetic structure. This finding further reinforces the high quality and reliability of our samples.
Collecting a large-scale phenotypes dataset of semen traits and genotypes of boars is challenging due to the high cost involved. However, a potential solution to this problem is to implement weighted GWAS, which helps reduce stratification and stabilize the solution by assigning weights to the residual variance [12]. This method effectively decreases the standard errors of the model parameters and increases the power of the tests. In this study, the lambda value, which indicates the extent of population stratification, was taken into account and was found to be close to 1 (ranging from 0.89 to 1.09). This suggests that reasonable consideration is given to population stratification within the three breeds. One significant advantage of the weighted GWAS lies in the mitigation of confounding effects related to population structure, which can otherwise lead to false positives or negatives in GWAS.
The GWAS results revealed many hits seem to be singletons, the GWAS regions represented by only one associated SNP. This finding is consistent with the studies conducted by Hering et al. [26] and Mei et al. [10], who also encountered a similar situation. This may be due to limitations in the 50k chip data, leading to the detection of fewer SNPs associated with the target trait. To avoid false positives for single significant SNPs, we combined RNA-seq data to analyze gene expression levels of candidate genes found in significant SNPs GWAS regions. Interestingly, it was found that the identified candidate genes associated with semen traits were highly expressed in testis tissue. The presence of a single significant SNP in the GWAS results does not necessarily indicate unreliable. It may be attributed to the limited number of SNPs available in the 50 K chip data compared to whole genome sequencing data, as noted by Wang et al. [28]. Using whole-genome sequencing data is more conducive to detecting a higher proportion of significant SNPs in GWAS.
Several candidate genes have been reported to be associated with spermatogenesis, testicular function, and male fertility in pig semen traits. However, only a limited number of the same candidate genes have been identified across different studies. The majority of candidate genes associated with semen traits are specific to certain breeds or populations, potentially due to differences in the degree of selection for semen traits [6]. Additionally, the frequency of the same gene can vary between populations, leading to significant SNPs identified in GWAS results for one population not necessarily being significant in other populations [29]. Nevertheless, the utilization of similar analytical methods may play a crucial role in the low reproducibility observed across different studies.
In this study, the candidate genes RSPH3 and DYNLT1 were found to be located in the same region (8.24~8.56 Mb) on chromosome 1. RSPH3 and DYNLT were identified as shared candidate genes of Landrace and Yorkshire, indicating a potential same QTL region between these two breeds. Mutations in the RSPH3 gene have been associated with primary ciliary dyskinesia in humans, a disease characterized by defects in axial filaments in mobile cilia and sperm flagella [30]. DYNLT1 encodes part of the motor complex and cytoplasmic dynamic protein and aberrant expression of DYNLT1 has been linked to male infertility in humans [31]. Additionally, GO and KEGG enrichment analysis revealed that the candidate genes identified in three pig breeds were involved in shared biological processes, which is consistent with findings from another study [8].
For Duroc, seven candidate genes associated with semen traits have been reported to be linked to the biological function of sperm. One of the candidate genes, CCDC38, is located on chromosome 5: 87.56–87.61 Mb. The protein encoded by the CCDC38 gene is considered a component of centromere protein in mammalian sperm cells and plays a crucial role in the nucleation of sperm ciliary axoneme [32]. DNAH10, located on chromosome 14 at 29.09–29.24 Mb, is in close proximity to SBNO1, located on chromosome 14 at 29.57–29.62 Mb. DNAH10 gene is a component of the outer arm and inner arm dynamic protein attached to the peripheral microtubule duplex, specifically the inner arm dynamic protein heavy chain. Biallelic mutations in this gene can lead to primary male infertility in humans and mice with asthenoteratozoospermia [33]. The expression of the SBNO1 gene is negatively correlated with human sperm motility [34]. Additionally, candidate genes CCDC62 and IFT81 are located on chromosome 14: 29.99–30.06 Mb and chromosome 14: 31.36–31.65 Mb, respectively. CCDC62 is involved in the response of cells to estrogen stimulation and the positive regulation of transcription by RNA polymerase II, activating estrogen receptor binding activity and nuclear receptor coactivator activity. It is associated with spermatogenesis defects and male infertility [35,36]. The protein encoded by IFT81, together with IFT74, forms a module of the intraflagellar transport complex B, which is responsible for the transport of tubulin within the cilium. This protein plays a crucial role in spermatogenesis, fertility, and ciliogenesis [37,38]. Another candidate gene, SPATA6, located on chromosome 6: 163.27–163.40 Mb, was also identified in a study conducted by Marques et al. [6]. The SPATA6 gene affects the expression of semen traits through biological processes such as cilium organization and assembly in mice and humans, particularly motile cilium assembly and spermatogenesis, which are active in sperm connecting pieces [39,40,41]. The GTSF1L is located on chromosome 17: 46.29–46.30 Mb and is specifically expressed in gonocytes and spermatids in mice [42].
For Landrace, five candidate genes associated with semen traits have been reported to be linked to the biological function of sperm. One of the candidate genes, DNAI2, is located on chromosome 12: 6.80–6.83 Mb and is part of the dynein complex of sperm flagella. It has been associated with sperm ciliary dyskinesia in humans [43], which is consistent with findings from studies conducted by Marques et al. [6] and Gao et al. [8]. The DNAI2 gene is highly expressed in the human testis, and its mutation leads to primary ciliary motility disorders, resulting in impaired sperm flagellar function, leading to decreased fertility in men [44,45,46]. Another candidate gene, METTL3, is located on chromosome 7: 77.66–77.69 Mb and plays an important role in regulating spermatogenesis and the initiation of meiosis in mice [47]. The candidate gene PNLDC1, located on chromosome 1: 7.56–7.58 Mb, has been found in the endoplasmic reticulum and is implicated in spermatogenic failure. PNLDC1 is necessary for meiosis and the development of male germ cells after meiosis in mice [48], affecting spermatogenesis and male fertility by modifying piRNA [49,50,51].
For Yorkshire, seven candidate genes associated with semen traits have been reported to be linked to the biological function of sperm. Among these candidate genes, IZUMO1R and PANX1, are located on chromosome 9: 26.44–26.52 Mb. IZUMO1R plays an important role in signal transduction during sperm–oocyte binding and the activation of sperm-binding site actin in mammals [52,53]. Mutations in this gene have been associated with in vitro fertilization failure in humans [54]. Homozygous variants in PANX1 have been found to cause oocyte death and female infertility [55]. LHX9 is located on chromosome 10: 20.74–20.76 Mb, belongs to the LIM homeobox gene family and encodes transcription factors involved in development. Inactivation of the LHX9 gene leads to gonadal hypoplasia in mice, indicating its crucial role in gonadal development [56,57]. Furthermore, DHH and CCDC65 are located on chromosome 5: 15.11–15.12 Mb and chromosome 5: 14.95–14.96 Mb, respectively. The DHH gene encodes a member of the Hedgehog family, which plays a critical role in regulating morphogenesis. Mutations in the DHH gene have been associated with partial gonadal hypoplasia with micronucleus polyneuropathy. DHH gene mutations affect male gonadal differentiation and neuromembrane development, with mild mutations impacting fertility and severe or multiple mutations leading to gonadal dysplasia [58,59,60]. The CCDC65 gene affects semen traits through biological processes involving the axoneme and ciliary basal body. It encodes a sperm tail protein that is highly expressed in adult testis, spermatocytes, and sperm cells. CCDC65 is a central hub for assembling nexin–dynein regulatory complexes, other cilia, and flagellar motility regulators in humans [61]. Mutations in CCDC65 alter cilia beating patterns and cause primary ciliary dyskinesia in humans [62].
The integration of GWAS and RNA-seq data offers a robust framework to elucidate the genetic architecture underlying complex traits. In the study, data from weighted GWAS and multi-tissue RNA-seq were combined to identify genetic variants and candidate genes across three pig breeds. The methodology for integrating these datasets hinges on leveraging eQTL analyses, which assess the impact of genetic variants on gene expression in specific tissues. For calculating differences between breeds, tissues, and breed-by-tissue interactions, we employed a mixed model framework. Genotypic data were encoded (e.g., AA, Aa, and aa as 0, 1, and 2, respectively) and analyzed alongside phenotypic data, such as de-regressed estimated breeding values. The RNA-seq data, representing expression levels across 34 tissues, were used to calculate transcripts per million values, which were then mapped to significant SNPs identified in the GWAS. Breed differences were captured by stratifying analyses per breed (Duroc, Landrace, and Yorkshire), while tissue-specific effects were assessed by examining eQTLs associated with significant SNPs in RNA-seq data from specific tissues, such as the testis. For example, SNP rs320928244, significant in Landrace for sperm count traits, was found to regulate the expression of the DYNLT1 gene specifically in testis tissue. This relationship was validated through linkage disequilibrium analysis and comparative genotype expression studies, revealing significant genotype-based expression differences. Similarly, for breed-by-tissue interactions, the study found overlapping eQTLs and significant SNPs for genes like DNAH10 in testis tissues across breeds, emphasizing both shared and distinct genetic regulation mechanisms. This integration method not only highlights breed-specific and shared candidate genes but also provides insights into the biological pathways active in semen traits, facilitating improved breeding strategies.
5. Conclusions
This study integrated a weighted GWAS and multi-tissue transcriptome analysis to identify genetic variants and candidate genes associated with semen traits in three pig breeds. The findings revealed that most candidate genes were linked to sperm flagellar assembly, including components of axonemal dynein arms and microtubules. Mutations in these candidate genes were predominantly associated with sperm motility defects and male infertility. The multi-tissue transcriptome analysis indicated high expression levels of the identified candidate genes associated with semen traits in the testis tissue of Duroc, Landrace, and Yorkshire. Furthermore, significant effects of three genotypes of the rs320928244 on the DYNLT1 gene expression in pig testis tissue. This study provides new insights into the genetic structure of semen traits in these three pig breeds, enabling the improvement of semen quality and yield through pig genome breeding.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ani15030438/s1, Figure S1: The distribution histogram with semen collection times of Duroc, Landrace, and Yorkshire; Figure S2: The distributions histogram of DEBVs phenotype for semen traits in three pig breeds; Figure S3: Quantile–quantile plots for GWAS results of semen traits in the Duroc, Landrace, and Yorkshire; Figure S4: The 34 tissues sample size of 5457 pig RNA-seq data from FarmGTEx; Figure S5: The expression levels of candidate genes associated with semen traits in 34 tissues; Table S1: Variance components of the semen traits of three pig breeds; Table S2: Descriptive statistics of DEBVs for the semen traits in three pig breeds; Table S3: All candidate genes in GWAS regions of semen traits in three pig breeds.
Author Contributions
Conceptualization, X.Z.; methodology, X.Z.; software, X.Z.; validation, Z.X. and Q.L.; formal analysis, X.Z. and Q.L.; investigation, Y.G.; resources, X.Q.; data curation, J.L.; writing—original draft preparation, X.Z. and Y.G.; writing—review and editing, X.Z., Q.L. and Y.G.; visualization, X.Z.; supervision, J.L. and S.X.; project administration, Y.G. and S.X.; funding acquisition, J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the earmarked fund for the China Agriculture Research System (CARS-35), and Guangdong Provincial Key R&D Program (2022B0202090002).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated and/or analyzed during the current study are not publicly available since the studied population consists of the nucleus herd of Guangdong Guyue Technology Co., Ltd., China, but are available from the corresponding author on reasonable request.
Acknowledgments
We thank the National Supercomputer Center in Guangzhou for its computing support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Knox, R.V. Artificial insemination in pigs today. Theriogenology 2016, 85, 83–93. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Q.; Liao, W.; Zhang, W.; Li, T.; Li, J.; Zhang, Z.; Huang, X.; Zhang, H. Identification of new candidate genes related to semen traits in Duroc pigs through weighted single-step GWAS. Animals 2023, 13, 365. [Google Scholar] [CrossRef]
- Nagai, R.; Kinukawa, M.; Watanabe, T.; Ogino, A.; Kurogi, K.; Adachi, K.; Satoh, M.; Uemoto, Y. Genome-wide detection of non-additive quantitative trait loci for semen production traits in beef and dairy bulls. Animal 2022, 16, 100472. [Google Scholar] [CrossRef]
- Dehghan, A. Genome-Wide Association Studies. Methods Mol. Biol. 2018, 1793, 37–49. [Google Scholar] [CrossRef]
- Diniz, D.B.; Lopes, M.S.; Broekhuijse, M.L.; Lopes, P.S.; Harlizius, B.; Guimaraes, S.E.; Duijvesteijn, N.; Knol, E.F.; Silva, F.F. A genome-wide association study reveals a novel candidate gene for sperm motility in pigs. Anim. Reprod. Sci. 2014, 151, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Marques, D.B.D.; Bastiaansen, J.W.M.; Broekhuijse, M.; Lopes, M.S.; Knol, E.F.; Harlizius, B.; Guimaraes, S.E.F.; Silva, F.F.; Lopes, P.S. Weighted single-step GWAS and gene network analysis reveal new candidate genes for semen traits in pigs. Genet. Sel. Evol. 2018, 50, 40. [Google Scholar] [CrossRef] [PubMed]
- Godia, M.; Reverter, A.; Gonzalez-Prendes, R.; Ramayo-Caldas, Y.; Castello, A.; Rodriguez-Gil, J.E.; Sanchez, A.; Clop, A. A systems biology framework integrating GWAS and RNA-seq to shed light on the molecular basis of sperm quality in swine. Genet. Sel. Evol. 2020, 52, 72. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Chen, Y.; Liu, X.; Zhao, Y.; Zhu, L.; Liu, A.; Jiang, W.; Peng, X.; Zhang, C.; Tang, Z.; et al. Weighted single-step GWAS identified candidate genes associated with semen traits in a Duroc boar population. BMC Genom. 2019, 20, 797. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, N.; Li, X.; El-Ashram, S.; Wang, Z.; Zhu, L.; Jiang, W.; Peng, X.; Zhang, C.; Chen, Y.; et al. Identifying candidate genes associated with sperm morphology abnormalities using weighted single-step GWAS in a Duroc boar population. Theriogenology 2020, 141, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Fu, C.; Sahana, G.; Chen, Y.; Yin, L.; Miao, Y.; Zhao, S.; Xiang, T. Identification of new semen trait-related candidate genes in Duroc boars through genome-wide association and weighted gene co-expression network analyses. J. Anim. Sci. 2021, 99, skab188. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Hu, L.; Brito, L.F.; Dou, J.; Sammad, A.; Chang, Y.; Ma, L.; Guo, G.; Liu, L.; Zhai, L.; et al. Weighted single-step GWAS and RNA sequencing reveals key candidate genes associated with physiological indicators of heat stress in Holstein cattle. J. Anim. Sci. Biotechnol. 2022, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; VanRaden, P.M.; Null, D.J.; O’Connell, J.R.; Cole, J.B. Major quantitative trait loci influencing milk production and conformation traits in Guernsey dairy cattle detected on Bos taurus autosome 19. J. Dairy. Sci. 2021, 104, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lim, B.; Cho, J.; Lee, S.; Dang, C.G.; Jeon, J.H.; Kim, J.M.; Lee, J. Genome-wide identification of candidate genes for milk production traits in Korean Holstein cattle. Animals 2021, 11, 1392. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef]
- Teng, J.; Gao, Y.; Yin, H.; Bai, Z.; Liu, S.; Zeng, H.; Bai, L.; Cai, Z.; Zhao, B.; Li, X.; et al. A compendium of genetic regulatory effects across pig tissues. Nat. Genet. 2024, 56, 112–123. [Google Scholar] [CrossRef]
- Marques, D.B.D.; Lopes, M.S.; Broekhuijse, M.; Guimaraes, S.E.F.; Knol, E.F.; Bastiaansen, J.W.M.; Silva, F.F.; Lopes, P.S. Genetic parameters for semen quality and quantity traits in five pig lines. J. Anim. Sci. 2017, 95, 4251–4259. [Google Scholar] [CrossRef]
- Wang, C.; Li, J.; Wei, H.; Zhou, Y.; Tan, J.; Sun, H.; Jiang, S.; Peng, J. Effects of feeding regimen on weight gain, semen characteristics, libido, and lameness in 170- to 250-kilogram Duroc boars. J. Anim. Sci. 2016, 94, 4666–4676. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G. Genome-Assisted Prediction of Quantitative Traits Using the R Package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Madsen, P.; Srensen, P.; Su, G.; Damgaard, L.H.; Labouriau, R.E. DMU—A package for analyzing multivariate mixed models. In Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, Brazil, 13–18 August 2006. [Google Scholar]
- VanRaden, P.M.; Van Tassell, C.P.; Wiggans, G.R.; Sonstegard, T.S.; Schnabel, R.D.; Taylor, J.F.; Schenkel, F.S. Invited review: Reliability of genomic predictions for North American Holstein bulls. J. Dairy. Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J. MMAP: Mixed Model Analysis for Pedigrees and Populations; University of Matyland School of Medicine: Baltimore, MD, USA, 2017. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Yang, J.; Weedon, M.N.; Purcell, S.; Lettre, G.; Estrada, K.; Willer, C.J.; Smith, A.V.; Ingelsson, E.; O’Connell, J.R.; Mangino, M.; et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 2011, 19, 807–812. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Hering, D.M.; Olenski, K.; Kaminski, S. Genome-wide association study for poor sperm motility in Holstein-Friesian bulls. Anim. Reprod. Sci. 2014, 146, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, B.; Wang, X.; Liu, X.; Zhang, Q.; Chen, Y. Estimation of genetic parameters and season effects for semen traits in three pig breeds of South China. J. Anim. Breed. Genet. 2019, 136, 183–189. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Shi, L.; Zhang, P.; Li, Y.; Li, M.; Tian, J.; Wang, L.; Zhao, F. GWAS of Reproductive Traits in Large White Pigs on Chip and Imputed Whole-Genome Sequencing Data. Int. J. Mol. Sci. 2022, 23, 13338. [Google Scholar] [CrossRef] [PubMed]
- Wu, C. Animals Genetics; Higher Education Press: Beijing, China, 2015; Volume 2, p. 382. [Google Scholar]
- Jeanson, L.; Copin, B.; Papon, J.F.; Dastot-Le Moal, F.; Duquesnoy, P.; Montantin, G.; Cadranel, J.; Corvol, H.; Coste, A.; Desir, J.; et al. RSPH3 mutations cause primary ciliary dyskinesia with central-complex defects and a near absence of radial spokes. Am. J. Hum. Genet. 2015, 97, 153–162. [Google Scholar] [CrossRef]
- Indu, S.; Sekhar, S.C.; Sengottaiyan, J.; Kumar, A.; Pillai, S.M.; Laloraya, M.; Kumar, P.G. Aberrant expression of dynein light chain 1 (DYNLT1) is associated with human male factor infertility. Mol. Cell Proteom. 2015, 14, 3185–3195. [Google Scholar] [CrossRef]
- Firat-Karalar, E.N.; Sante, J.; Elliott, S.; Stearns, T. Proteomic analysis of mammalian sperm cells identifies new components of the centrosome. J. Cell Sci. 2014, 127 Pt 19, 4128–4133. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Cong, J.; Zhang, Q.; He, X.; Zheng, R.; Yang, X.; Gao, Y.; Wu, H.; Lv, M.; Gu, Y.; et al. Bi-allelic mutations of DNAH10 cause primary male infertility with asthenoteratozoospermia in humans and mice. Am. J. Hum. Genet. 2021, 108, 1466–1477. [Google Scholar] [CrossRef]
- Freitas, M.J.; Silva, J.V.; Brothag, C.; Regadas-Correia, B.; Fardilha, M.; Vijayaraghavan, S. Isoform-specific GSK3A activity is negatively correlated with human sperm motility. Mol. Hum. Reprod. 2019, 25, 171–183. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Lin, S.; Yang, B.; Huang, W.; Wu, H.; Chen, Y.; Yang, L.; Luo, M.; Guo, H.; et al. A nonsense mutation in Ccdc62 gene is responsible for spermiogenesis defects and male infertility in repro29/repro29 mice. Biol. Reprod. 2017, 96, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Oud, M.S.; Okutman, O.; Hendricks, L.A.J.; de Vries, P.F.; Houston, B.J.; Vissers, L.; O’Bryan, M.K.; Ramos, L.; Chemes, H.E.; Viville, S.; et al. Exome sequencing reveals novel causes as well as new candidate genes for human globozoospermia. Hum. Reprod. 2020, 35, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhou, T.; Huang, Q.; Zhang, S.; Li, W.; Zhang, L.; Hess, R.A.; Pazour, G.J.; Zhang, Z. Intraflagellar transport protein 74 is essential for spermatogenesis and male fertility in micedagger. Biol. Reprod. 2019, 101, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Yuan, S.; Quan, C.; Huang, Q.; Zhou, Q.; Yap, Y.; Shi, L.; Zhang, D.; Guest, T.; Li, W.; et al. The essential role of intraflagellar transport protein IFT81 in male mice spermiogenesis and fertility. Am. J. Physiol. Cell Physiol. 2020, 318, C1092–C1106. [Google Scholar] [CrossRef] [PubMed]
- Yamano, Y.; Ohyama, K.; Sano, T.; Ohta, M.; Shimada, A.; Hirakawa, Y.; Sugimoto, M.; Morishima, I. A novel spermatogenesis-related factor-1 gene expressed in maturing rat testis. Biochem. Biophys. Res. Commun. 2001, 289, 888–893. [Google Scholar] [CrossRef]
- Yuan, S.; Stratton, C.J.; Bao, J.; Zheng, H.; Bhetwal, B.P.; Yanagimachi, R.; Yan, W. Spata6 is required for normal assembly of the sperm connecting piece and tight head-tail conjunction. Proc. Natl. Acad. Sci. USA 2015, 112, E430–E439. [Google Scholar] [CrossRef] [PubMed]
- Sujit, K.M.; Singh, V.; Trivedi, S.; Singh, K.; Gupta, G.; Rajender, S. Increased DNA methylation in the spermatogenesis-associated (SPATA) genes correlates with infertility. Andrology 2020, 8, 602–609. [Google Scholar] [CrossRef]
- Takemoto, N.; Yoshimura, T.; Miyazaki, S.; Tashiro, F.; Miyazaki, J. Gtsf1l and Gtsf2 are specifically expressed in gonocytes and spermatids but are not essential for spermatogenesis. PLoS ONE 2016, 11, e0150390. [Google Scholar] [CrossRef]
- Olcese, C.; Patel, M.P.; Shoemark, A.; Kiviluoto, S.; Legendre, M.; Williams, H.J.; Vaughan, C.K.; Hayward, J.; Goldenberg, A.; Emes, R.D.; et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat. Commun. 2017, 8, 14279. [Google Scholar] [CrossRef] [PubMed]
- Pennarun, G.; Chapelin, C.; Escudier, E.; Bridoux, A.-M.; Dastot, F.; Cacheux, V.; Goossens, M.; Amselem, S.; Duriez, B. The human dynein intermediate chain 2 gene (DNAI2): Cloning, mapping, expression pattern, and evaluation as a candidate for primary ciliary dyskinesia. Hum Genet. 2000, 107, 642–649. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Fenske, L.; Mussaffi, H.; Horvath, J.; Fliegauf, M.; Kuhl, H.; Baktai, G.; Peterffy, E.; Chodhari, R.; et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008, 83, 547–558. [Google Scholar] [CrossRef]
- Al-Mutairi, D.A.; Alsabah, B.H.; Alkhaledi, B.A.; Pennekamp, P.; Omran, H. Identification of a novel founder variant in DNAI2 cause primary ciliary dyskinesia in five consanguineous families derived from a single tribe descendant of Arabian Peninsula. Front. Genet. 2022, 13, 1017280. [Google Scholar] [CrossRef]
- Xu, K.; Yang, Y.; Feng, G.H.; Sun, B.F.; Chen, J.Q.; Li, Y.F.; Chen, Y.S.; Zhang, X.X.; Wang, C.X.; Jiang, L.Y.; et al. Mettl3-mediated m(6)A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017, 27, 1100–1114. [Google Scholar] [CrossRef]
- Nishimura, T.; Nagamori, I.; Nakatani, T.; Izumi, N.; Tomari, Y.; Kuramochi-Miyagawa, S.; Nakano, T. PNLDC1, mouse pre-piRNA Trimmer, is required for meiotic and post-meiotic male germ cell development. EMBO Rep. 2018, 19, e44957. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Liu, J.; Dong, K.; Midic, U.; Hess, R.A.; Xie, H.; Demireva, E.Y.; Chen, C. PNLDC1 is essential for piRNA 3′ end trimming and transposon silencing during spermatogenesis in mice. Nat. Commun. 2017, 8, 819. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, R.; Cui, Y.; Zhu, Z.; Zhang, Y.; Wu, H.; Zheng, B.; Yue, Q.; Bai, S.; Zeng, W.; et al. An essential role for PNLDC1 in piRNA 3′ end trimming and male fertility in mice. Cell Res. 2017, 27, 1392–1396. [Google Scholar] [CrossRef]
- Bronkhorst, A.W.; Ketting, R.F. Trimming it short: PNLDC1 is required for piRNA maturation during mouse spermatogenesis. EMBO Rep. 2018, 19, e45824. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N. Novel insights into the molecular mechanism of sperm-egg fusion via IZUMO1. J. Plant Res. 2017, 130, 475–478. [Google Scholar] [CrossRef]
- Wang, H.; Hong, X.; Kinsey, W.H. Sperm-oocyte signaling: The role of IZUMO1R and CD9 in PTK2B activation and actin remodeling at the sperm binding sitedagger. Biol. Reprod. 2021, 104, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhao, H.; Chen, T.; Tian, Y.; Li, M.; Wu, K.; Bian, Y.; Su, S.; Cao, Y.; Ning, Y.; et al. Mutational analysis of IZUMO1R in women with fertilization failure and polyspermy after in vitro fertilization. J. Assist. Reprod. Genet. 2018, 35, 539–544. [Google Scholar] [CrossRef]
- Wang, W.; Qu, R.; Dou, Q.; Wu, F.; Wang, W.; Chen, B.; Mu, J.; Zhang, Z.; Zhao, L.; Zhou, Z.; et al. Homozygous variants in PANX1 cause human oocyte death and female infertility. Eur. J. Hum. Genet. 2021, 29, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Ottolenghi, C.; Moreira-Filho, C.; Mendonça, B.B.; Barbieri, M.; Fellous, M.; Berkovitz, G.D.; McElreavey, K. Absence of mutations involving the LIM homeobox domain gene LHX9 in 46,XY gonadal agenesis and dysgenesis. J. Clin. Endocrinol. Metab. 2001, 86, 2465–2469. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Singh, D.; Modi, D. LIM Homeodomain (LIM-HD) genes and their co-regulators in developing reproductive system and disorders of sex development. Sex. Dev. 2021, 16, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Tajouri, A.; Kharrat, M.; Hizem, S.; Zaghdoudi, H.; M’Rad, R.; Simic-Schleicher, G.; Kaiser, F.J.; Hiort, O.; Werner, R. In vitro functional characterization of the novel DHH mutations p.(Asn337Lysfs*24) and p.(Glu212Lys) associated with gonadal dysgenesis. Hum. Mutat. 2018, 39, 2097–2109. [Google Scholar] [CrossRef] [PubMed]
- Elzaiat, M.; Flatters, D.; Sierra-Diaz, D.C.; Legois, B.; Laissue, P.; Veitia, R.A. DHH pathogenic variants involved in 46,XY disorders of sex development differentially impact protein self-cleavage and structural conformation. Hum. Genet. 2020, 139, 1455–1470. [Google Scholar] [CrossRef]
- Mehta, P.; Singh, P.; Gupta, N.J.; Sankhwar, S.N.; Chakravarty, B.; Thangaraj, K.; Rajender, S. Mutations in the desert hedgehog (DHH) gene in the disorders of sexual differentiation and male infertility. J. Assist. Reprod. Genet. 2021, 38, 1871–1878. [Google Scholar] [CrossRef]
- Bower, R.; Tritschler, D.; Mills, K.V.; Heuser, T.; Nicastro, D.; Porter, M.E. DRC2/CCDC65 is a central hub for assembly of the nexin-dynein regulatory complex and other regulators of ciliary and flagellar motility. Mol. Biol. Cell 2018, 29, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Brody, S.L.; Ferkol, T.W.; Shoseyov, D.; Wasserman, M.G.; Ta-shma, A.; Wilson, K.S.; Bayly, P.V.; Amirav, I.; Cohen-Cymberknoh, M.; et al. CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS ONE 2013, 8, e72299. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).