
Genetic Comparison and Selection of Reproductive and Growth-Related Traits in Qinchuan Cattle and Two Belgian Cattle Breeds

 ,
,  , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genotyping Data Sources
2.2. SNP Chip Quality Control and Filling
2.3. LD Calculation Method
2.4. Estimation of Effective Group Size
2.5. Diversity of Population Structure
2.6. Fixation Index
2.7. Nucleotide Diversity (θπ Ratio) Analysis
2.8. Enrichment Analysis of Candidate Genes
3. Results
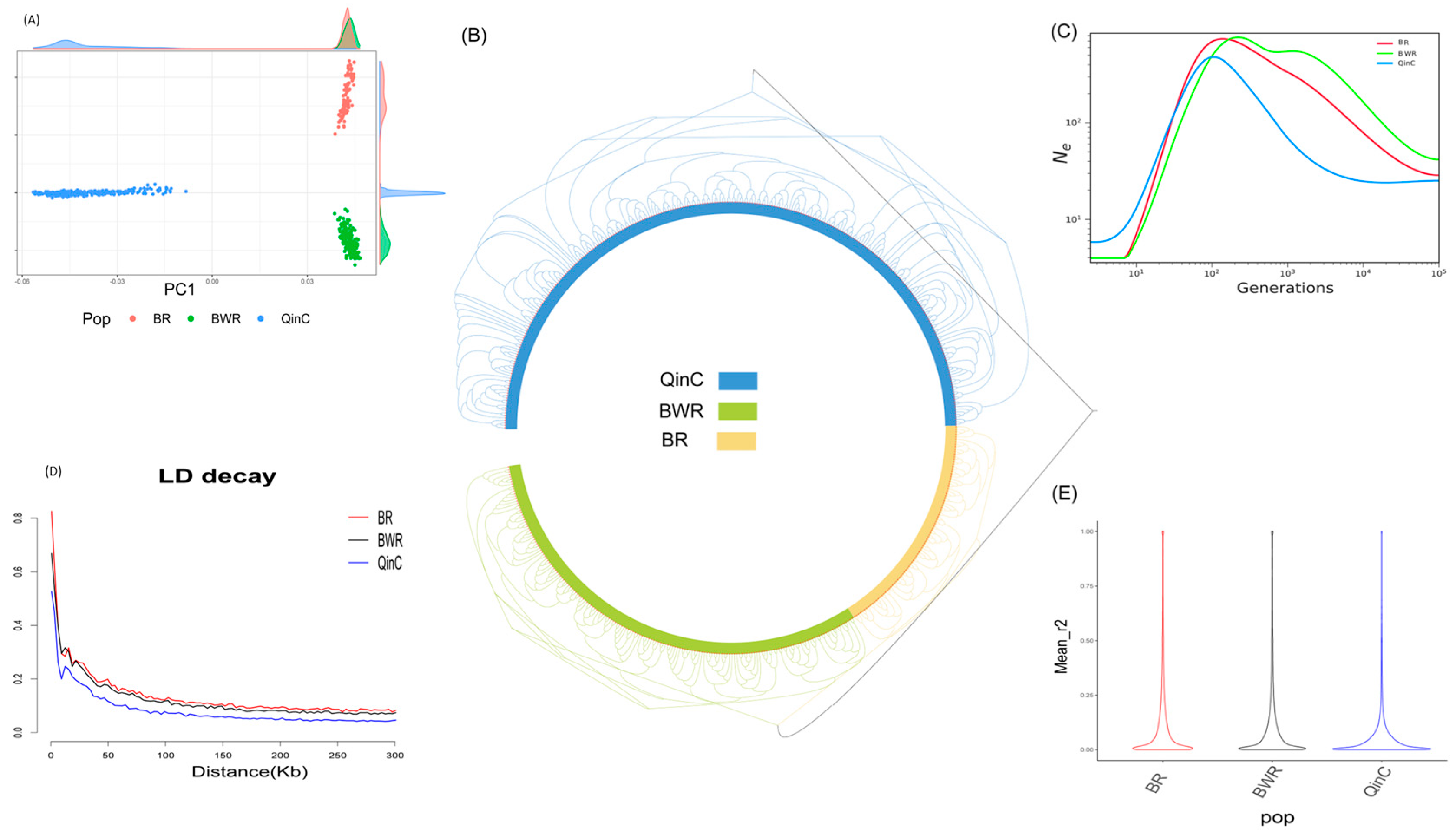
3.1. Population Structure
3.2. Selective Sweeps
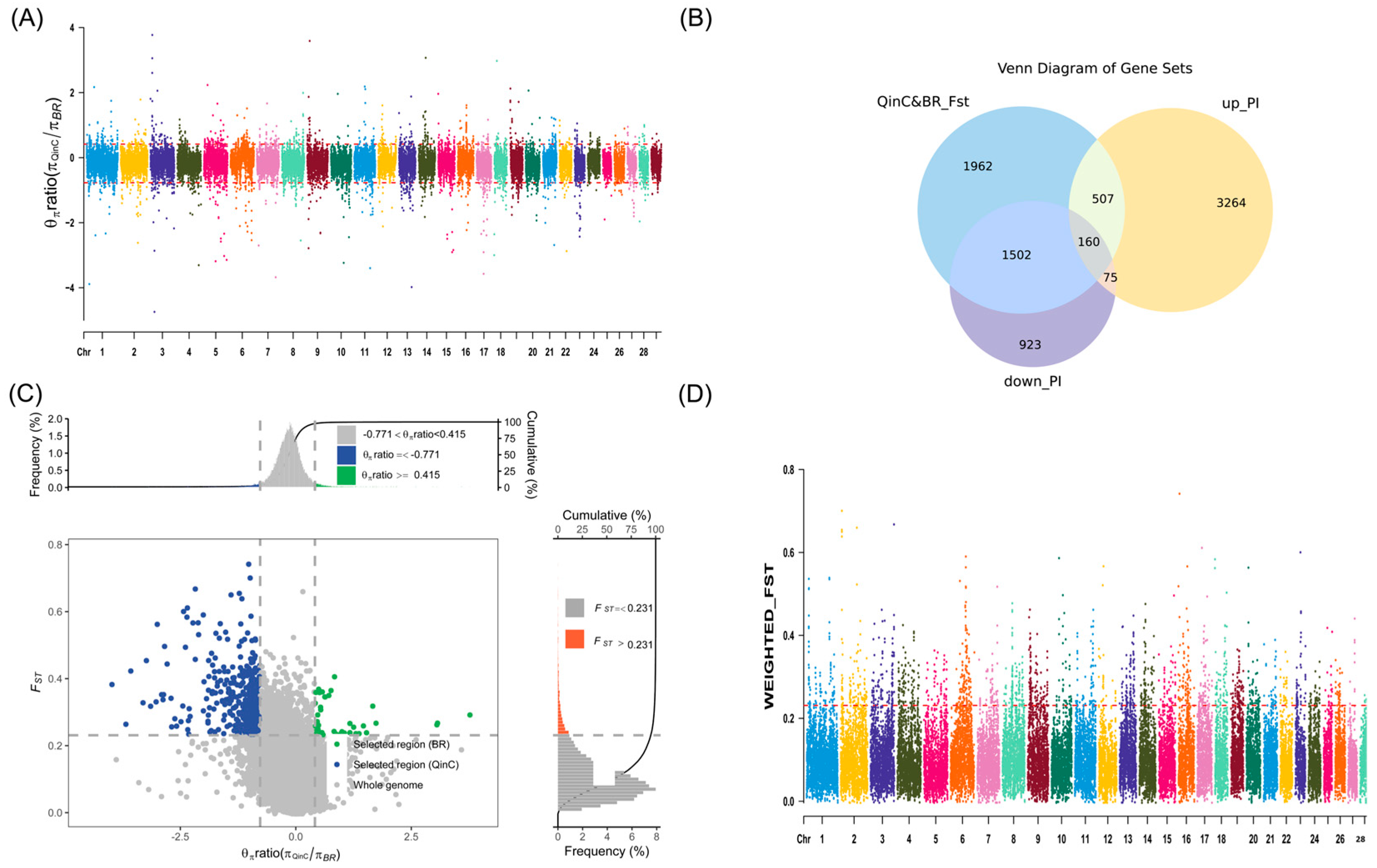
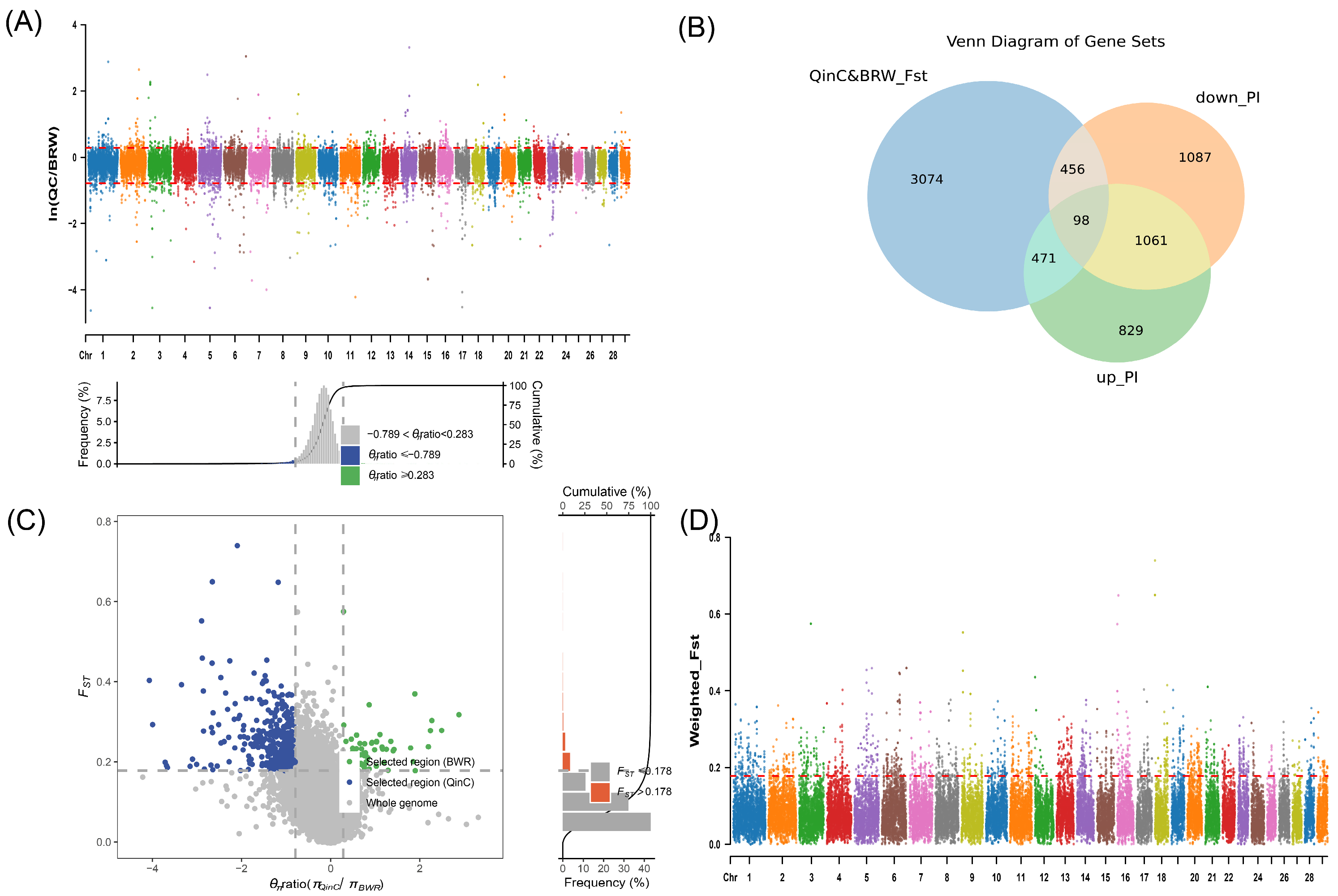
3.2.1. Fixation Index Results
3.2.2. Nucleotide Diversity (θπ Ratio) Analysis
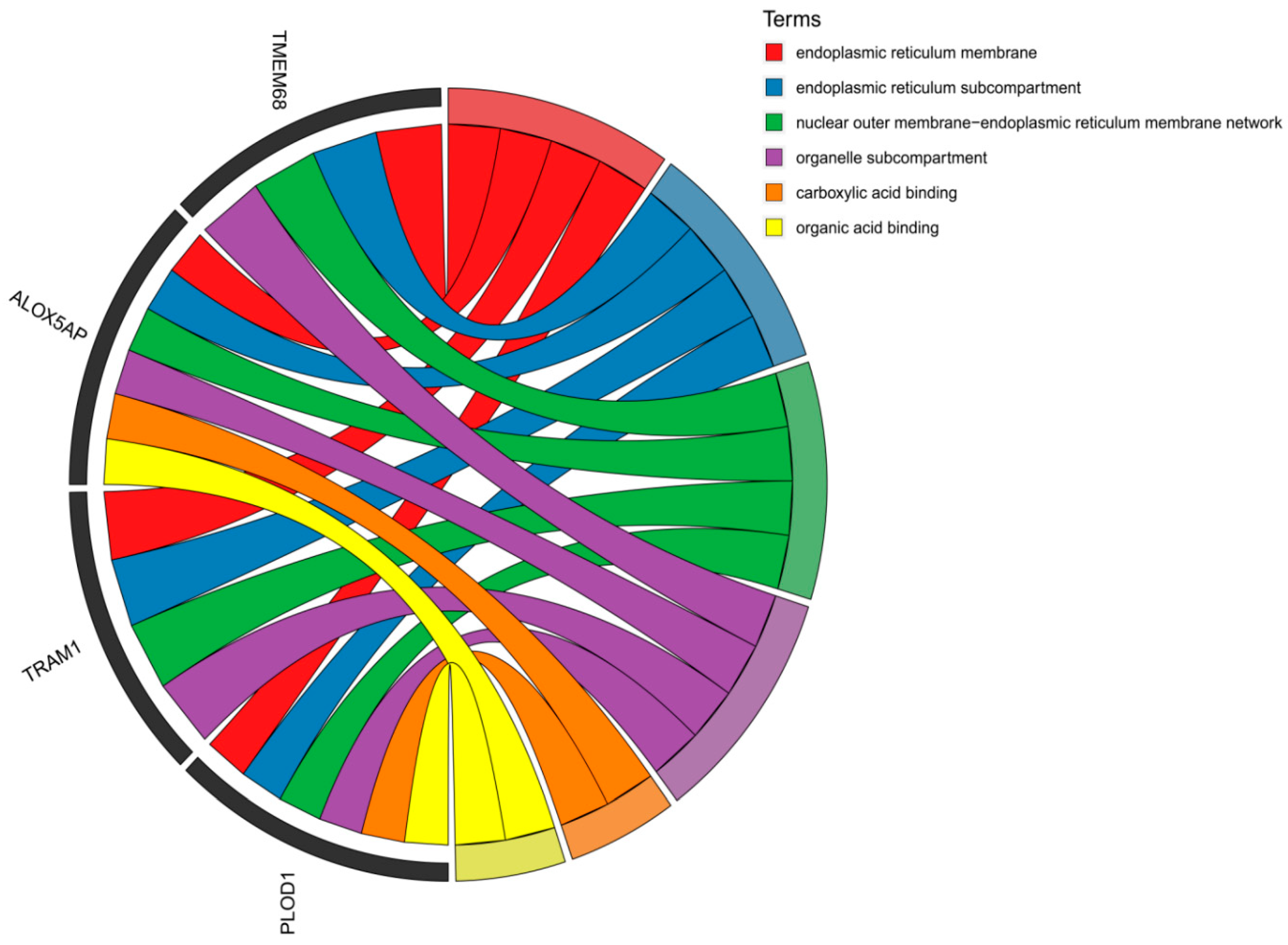
3.2.3. Enrichment Analysis of Candidate Genes Results
4. Discussion
4.1. Analysis of Population Structure
4.2. Reproductive Performance-Related Genes
4.3. Analysis of Genes Related to Growth, Development, and Meat Production
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. Genomics Confirm an Alarming Status of the Genetic Diversity of Belgian Red and Belgian White Red Cattle. Animals 2021, 11, 3574. [Google Scholar] [CrossRef]
- Farquharson, K.A.; Hogg, C.J.; Grueber, C.E. A meta-analysis of birth-origin effects on reproduction in diverse captive environments. Nat. Commun. 2018, 9, 1055. [Google Scholar] [CrossRef]
- Neumann, G.B.; Korkuć, P.; Arends, D.; Wolf, M.J.; May, K.; König, S.; Brockmann, G.A. Genomic diversity and relationship analyses of endangered German Black Pied cattle (DSN) to 68 other taurine breeds based on whole-genome sequencing. Front. Genet. 2023, 13, 993959. [Google Scholar] [CrossRef]
- Ma, J.; Fan, A.-P.; Wang, W.-S.; Zhang, J.-C.; Jiang, X.-J.; Ma, R.-J.; Jia, S.-Q.; Liu, F.; Lei, C.-C.; Huang, Y.-Z. Analysis of genetic diversity and genetic structure of Qinchuan cattle conservation population using whole-genome resequencing. Yi Chuan 2023, 45, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; He, S.; Chen, L.; Li, W.; Di, J.; Liu, M. Estimates of linkage disequilibrium and effective population sizes in Chinese Merino (Xinjiang type) sheep by genome-wide SNPs. Genes. Genom. 2017, 39, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Tenesa, A.; Navarro, P.; Hayes, B.J.; Duffy, D.L.; Clarke, G.M.; Goddard, M.E.; Visscher, P.M. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007, 17, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Estimation of effective population sizes from data on genetic markers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-L.; Zhang, J.; Tuersuntuoheti, M.; Chang, Q.; Liu, S. Population structure, genetic diversity and prolificacy in pishan red sheep under an extreme desert environment. Front. Genet. 2023, 14, 1092066. [Google Scholar] [CrossRef]
- Card, C.; Anderson, E.J.; Zamberlan, S.; Kaproth, M.; Karin, K.E.; Sartini, B.L. Selection signatures of QinChuan cattle based on whole-genome sequences. Anim. Biotechnol. 2023, 34, 1483–1491. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G. Estimation of linkage disequilibrium in randomly mating populations. Heredity 1974, 33, 229–239. [Google Scholar] [CrossRef]
- Terhorst, J.; Kamm, J.A.; Song, Y.S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 2017, 49, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Lenstra, J.A.; Zhang, S.; Liu, W.; Liu, J. Evolution and domestication of the Bovini species. Anim. Genet. 2020, 51, 637–657. [Google Scholar] [CrossRef] [PubMed]
- Mohammaddiyeh, M.E.T.K.; Rafat, S.A.; Shodja, J.; Javanmard, A.; Esfandyari, H. Selective genotyping to implement genomic selection in beef cattle breeding. Front. Genet. 2023, 14, 1083106. [Google Scholar] [CrossRef]
- Fiems, L.; De Campeneere, S.; Van Caelenbergh, W.; De Boever, J.; Vanacker, J. Carcass and meat quality in double-muscled Belgian Blue bulls and cows. Meat Sci. 2003, 63, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Marian, L.; Withoeft, J.A.; Costa, L.d.S.; Ribeiro, L.R.; Melo, I.C.; Alves, R.S.; Baumbach, L.F.; Pinto, M.G.L.; Snak, A.; Miletti, L.C.; et al. Causes of fetal death in the Flemish cattle herd in Brazil. Vet. World 2023, 16, 766–772. [Google Scholar] [CrossRef] [PubMed]
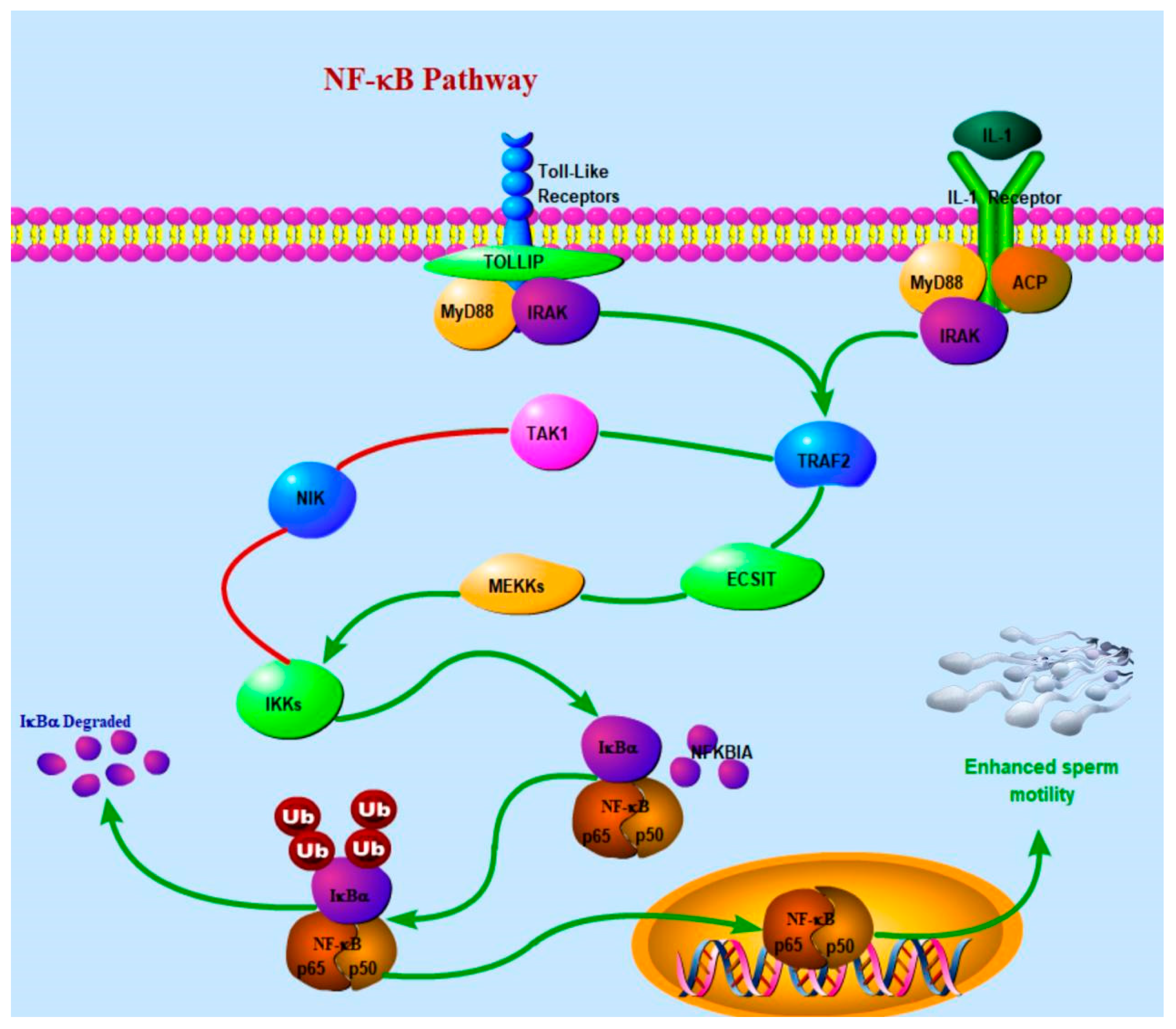
- Li, Y.; Hu, Y.; Wang, Z.; Lu, T.; Yang, Y.; Diao, H.; Zheng, X.; Xie, C.; Zhang, P.; Zhang, X.; et al. IKBA phosphorylation governs human sperm motility through ACC-mediated fatty acid beta-oxidation. Commun. Biol. 2023, 6, 323. [Google Scholar] [CrossRef]
- Chear, C.T.; El Farran, B.A.K.; Sham, M.; Ramalingam, K.; Noh, L.M.; Ismail, I.H.; Chiow, M.Y.; Baharin, M.F.; Ripen, A.M.; Bin Mohamad, S. A Novel De Novo NFKBIA Missense Mutation Associated to Ectodermal Dysplasia with Dysgammaglobulinemia. Genes 2022, 13, 1900. [Google Scholar] [CrossRef]
- Hens, J.R.; Dann, P.; Zhang, J.-P.; Harris, S.; Robinson, G.W.; Wysolmerski, J. BMP4 and PTHrP interact to stimulate ductal outgrowth during embryonic mammary development and to inhibit hair follicle induction. Development 2007, 134, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Tian, T.; Gao, P.; Fang, X.; Jiang, W.; Li, Z.; Tang, K.; Ouyang, P.; Li, L. Unveiling potential therapeutic targets for diabetes-induced frozen shoulder through Mendelian randomization analysis of the human plasma proteome. BMJ Open Diabetes Res. Care 2024, 12, e003966. [Google Scholar] [CrossRef]
- Molin, A.N.; Contentin, R.; Angelozzi, M.; Karvande, A.; Kc, R.; Haseeb, A.; Voskamp, C.; de Charleroy, C.; Lefebvre, V. Skeletal growth is enhanced by a shared role for SOX8 and SOX9 in promoting reserve chondrocyte commitment to columnar proliferation. Proc. Natl. Acad. Sci. USA 2024, 121, e2316969121. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Yang, Y.; Long, F.; Sun, C. rs294775 is a cis-regulatory SNP for human UGT2B10. Clin. Exp. Pharmacol. Physiol. 2018, 45, 614–616. [Google Scholar] [CrossRef]
- Haakensen, V.D.; Biong, M.; Lingjærde, O.C.; Holmen, M.M.; Frantzen, J.O.; Chen, Y.; Navjord, D.; Romundstad, L.; Lüders, T.; Bukholm, I.K.; et al. Expression levels of uridine 5'-diphospho-glucuronosyltransferase genes in breast tissue from healthy women are associated with mammographic density. Breast Cancer Res. 2010, 12, R65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Bosch, M.A.; Rønnekleiv, O.K.; Kelly, M.J. Kisspeptin activation of TRPC4 channels in female GnRH neurons requires PIP2 depletion and cSrc kinase activation. Endocrinology 2013, 154, 2772–2783. [Google Scholar] [CrossRef]
- Freichel, M.; Tsvilovskyy, V.; Camacho-Londoño, J.E. TRPC4- and TRPC4-containing channels. Handb. Exp. Pharmacol. 2014, 222, 85–128. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Z. TRPC4 ion channel regulations by small-molecular inhibitors and calmodulin. Cell Calcium. 2021, 95, 102361. [Google Scholar] [CrossRef]
- Plant, T.D.; Schaefer, M. TRPC4 and TRPC5: Receptor-operated Ca2+-permeable nonselective cation channels. Cell Calcium. 2003, 33, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Brunetti, V.; Perna, A.; Guerra, G.; Soda, T.; Berra-Romani, R. The Molecular Heterogeneity of Store-Operated Ca2+ Entry in Vascular Endothelial Cells: The Different roles of Orai1 and TRPC1/TRPC4 Channels in the Transition from Ca2+-Selective to Non-Selective Cation Currents. Int. J. Mol. Sci. 2023, 24, 3259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Bu, D.; Riboni, M.V.; Khan, M.; Graugnard, D.; Luo, J.; Cardoso, F.; Loor, J. Prepartal dietary energy level affects peripartal bovine blood neutrophil metabolic, antioxidant, and inflammatory gene expression. J. Dairy. Sci. 2015, 98, 5492–5505. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Q.; Kong, L.; Xia, F.; Yan, H.; Zhu, Y.; Mao, P. Proteomic and Physiological Analysis of the Response of Oat (Avena sativa) Seeds to Heat Stress under Different Moisture Conditions. Front. Plant Sci. 2016, 7, 896. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I.; Kitakaze, M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ. Res. 2010, 107, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Xu, R. Roles of PLODs in Collagen Synthesis and Cancer Progression. Front. Cell Dev. Biol. 2018, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Hashidate-Yoshida, T.; Shimizu, T.; Shindou, H. Profiling of fatty acid metabolism in the dorsal root ganglion after peripheral nerve injury. Front. Pain. Res. 2022, 3, 948689. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Term | Pvalue | Gene |
|---|---|---|---|
| Growth and development | GO:0061035~regulation of cartilage development | 0.021454098 | PTHLH |
| GO:0002062~chondrocyte differentiation | 0.044240533 | PTHLH | |
| GO:0030282~bone mineralization | 0.056316761 | PTHLH | |
| GO:0061448~connective tissue development | 0.091748173 | PTHLH | |
| bta00040~Pentose and glucuronate interconversions | 9.53677E-05 | UGT2B10 | |
| Reproductive correlation | GO:0045596~negative regulation of cell differentiation | 0.163777049 | PTHLH |
| bta04929~GnRH secretion | 0.159818885 | TRPC4 | |
| bta04917~Prolactin signaling pathway | 0.203839237 | GCK | |
| bta04926~Relaxin signaling pathway | 0.298730333 | NFKBIA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Niu, P.; Wang, X.; Huang, F.; Wang, J.; Qu, H.; Han, C.; Gao, Q. Genetic Comparison and Selection of Reproductive and Growth-Related Traits in Qinchuan Cattle and Two Belgian Cattle Breeds. Animals 2025, 15, 608. https://doi.org/10.3390/ani15040608
Li X, Niu P, Wang X, Huang F, Wang J, Qu H, Han C, Gao Q. Genetic Comparison and Selection of Reproductive and Growth-Related Traits in Qinchuan Cattle and Two Belgian Cattle Breeds. Animals. 2025; 15(4):608. https://doi.org/10.3390/ani15040608
Chicago/Turabian StyleLi, Xiaopeng, Peng Niu, Xueyan Wang, Fei Huang, Jieru Wang, Huimin Qu, Chunmei Han, and Qinghua Gao. 2025. "Genetic Comparison and Selection of Reproductive and Growth-Related Traits in Qinchuan Cattle and Two Belgian Cattle Breeds" Animals 15, no. 4: 608. https://doi.org/10.3390/ani15040608
APA StyleLi, X., Niu, P., Wang, X., Huang, F., Wang, J., Qu, H., Han, C., & Gao, Q. (2025). Genetic Comparison and Selection of Reproductive and Growth-Related Traits in Qinchuan Cattle and Two Belgian Cattle Breeds. Animals, 15(4), 608. https://doi.org/10.3390/ani15040608