1. Introduction
Stress is the normal physiological response of the body to environmental or psychological changes, and numerous studies have reported that chronic stress induces physiological and pathophysiological damage [
1]. Cold exposure is a common problem as a stress to humans who live or work in extreme environments, and this exposure can induce cold stress and a large amount of studies have indicated cold stress have a negative influence on organism [
2,
3]. Many studies about stress have reported that stress activates the hypothalamic-pituitary-adrenal (HPA) axis response, and releases stress hormones such as corticosterones (CORT) to resist the negative effects of stress. However, chronic stimulation from stress can induce over-activation of the HPA axis and cause CORT secretory abnormalities [
4]. The hippocampus is the first target tissue of CORT, and chronic stress causing excessive CORT may result in irreversible damage to the hippocampus [
5,
6]. A previous study has demonstrated that chronic stress could induce oxidative stress, neuronal apoptosis, and other nonspecific reactions in the brain of mice, which influence the CA1 and CA3 region functions of the hippocampus manifested as emotion, memory, adaptability, and learning-ability dysfunctions [
5]. In our previous study, neuroinflammation [
7,
8], increased oxidation, and neuronal loss following acute and chronic cold exposure in the CA1 and CA3 regions of mice hippocampus were reported; as we know, CA1 and CA3 are the key regions of hippocampus, however, the underlying mechanism is not yet known, and whether the above phenomenon is associated with an excess of CORT has not been established.
Autophagy is the primary endogenous process of eradicating damaged cell organelles or unused proteins. Autophagy can be induced by internal and external factors including nutrient and energy deficiencies, oxidation stress, and mitochondrial damage [
2,
3,
9]. Some studies have reported that autophagy is active following stimulation by stress during tissue repair [
10]. In our previous study, the phenomenon of oxidation stress induced by excess CORT was shown to occur in the hippocampus of mice after cold exposure. However, whether there is an association between autophagy and cold exposure has not yet been determined.
Chronic cold stress has previously been reported to break the homeostasis of the hippocampus, which may be a major factor in neurodegenerative diseases, but the mechanism of the homeostasis imbalance in the hippocampus following cold exposure is not understood. In the present study, we characterized the phenomenon of autophagy in the hippocampus of mice following cold exposure and identified the mechanism, as well as consequences, of autophagy and CORT exposure in hippocampal HT-22 cells after CORT exposure.
2. Materials and Methods
2.1. Animals and the Experimental Design
Adolescent male and female C57BL/6 mice (5-weeks-old; 22–24 g) were purchased from Charles River (Beijing, China). The conditions of cold exposure have been previously described [
11]. Each group was pre-fed in a climatic chamber at an ambient temperature of 24 ± 2 °C and 40% relative humidity, under a 12/12 h light/dark cycle (lights on from 8:00 am to 8:00 pm), with free access to food and water for 1 week. The animals were divided into four groups: the cold exposure male (CEM) group; the cold exposure female (CEF) group; the room temperature male (RTM) group; the room temperature female (RTF) group. The CEM and CEF groups were transferred to a climatic chamber at 4 °C for 3 h per day, and then back to room temperature between the hours of 8:00 am and 8:00 pm. The process of chronic cold exposure continued for 7 days. All experimental procedures were approved by the Management Committee of the Experimental Animal Center of Heilongjiang Bayi Agricultural University.
2.2. Hippocampus Tissue Protein Extraction
The method to extract the protein was referenced to our previous study [
7]. In short, total hippocampal protein was extracted with 150 μL radio immunoprecipitation assay (RIPA) buffer (Beyotime, Hangzhou, China) containing 15 mM
phenylmethylsulfonyl fluoride (PMSF; Beyotime, Hangzhou, China). The protein concentration was determined using the Enhanced BCA Protein Assay Kit (Beyotime, Hangzhou, China) according to the manufacturer’s instructions. The samples were stored at −80 °C for western blot analysis.
2.3. Cell Culture and Reagents
Mouse hippocampal HT22 cells were a generous gift from Professor Liu (College of Veterinary Medicine, Jilin University, Jilin, China) and maintained in the recommended culture conditions. The cells were maintained in Dulbecco’s Modified Essential Medium supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) at 37 °C in a 5% CO2 humidified incubator. The cells were grown in a monolayer and routinely passaged two or three times a week.
2.4. CORT and RU486 Treatment
CORT and RU486 (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in dimethyl sulfoxide (DMSO) (Solarbio, Beijing, China). HT-22 cells were incubated with or without RU486 for 1 h, then the cells was treated with 400 μM CORT to build the model of CORT exposure referenced from previous studies [
12], for 3 h to obtain an excess CORT model.
2.5. Annexin V-FITC/propidium Iodide (PI) Staining
After CORT incubated, HT22 cells were digested with trypsin and collected. To quantitate cell death, the cells were stained with fluorescein isothiocyanate (FITC)-labeled propidiumiodide (PI) and annexin V (Beyotime) for 30 min. Fluorescence labeling was analyzed by two-color flow cytometry. Annexin V and PI emissions were detected in the FL1 and FL2 channels of a flow cytometer (CytoFLEX FCM; Beckman, Brea, CA, USA) using emission filters of 488 and 532 nm, respectively reference from our previous study [
13].
2.6. Adenosine Triphosphate (ATP)/Adenosine diphosphate (ADP)/Adenosine Monophosphate (AMP) Analyses
After CORT treatment, the levels of ATP/ADP/AMP of HT22 cells were determined by high-performance liquid chromatography using a Sepax Bio-C
18 column (4.6 mm i.d. × 250 mm; 5 μm and 200 angstrom beads) and a UV detector at a wavelength of 254 nm (bandwidth:16 nm) as previously described [
14].
2.7. Measurement of the Mitochondrial Membrane Potential
The mitochondrial membrane potential (∆Ψm) of the cells after CORT treated was measured. In a nutshell, HT22 cells were maintained in 20 mm confocal petri dishes (NEST, Jiangsu, China) until cell attachment and use CORT treatment, the ∆Ψm was detected using a mitochondrial membrane potential assay kit, according to the manufacturer’s instructions, and then the cells were viewed using a laser scanning confocal microscope (TCS SP2, Leica, Wetzlar, Germany).
2.8. Monodansylcadaverine (MDC) Staining
HT22 cells were maintained in 20 mm confocal petri dishes (NEST) and after CORT treatment, autophagy in HT22 cells was assessed after staining with an MDC assay kit following the manufacturer’s instructions (Solarbio, Beijing, China). All cells were viewed using a laser scanning confocal microscope.
2.9. Cell Protein Extraction
Total cell proteins were extracted from cell samples treated with 400 μM CORT using 100 μL RIPA buffer containing 10 mM PMSF. The samples were stored at −80 °C for western blot analysis. Protein concentration was determined using the Enhanced BCA Protein Assay Kit according to the manufacturer’s instructions.
2.10. Cell Nuclear Protein Extraction
Nuclear proteins were extracted from the cell sample treated with 400 μM CORT using the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Hangzhou, China), and the protein concentration was determined using the Enhanced BCA Protein Assay Kit according to the manufacturer’s instructions.
2.11. Western Blot Analysis
Approximately 30 μg of hippocampus and cell total protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (0.22 μm and 0.45 μm; Millipore, Darmstadt, Germany). The membranes were blocked in 5% nonfat milk in TBST (Tris-HCl, NaCl, and Tween 20) for 1 h at room temperature, then incubated overnight at 4°C with the following primary antibodies: glucocorticoid receptor (GR) (#24050-1-AP, 1:6000), cytochrome C (#10993-1-AP, 1:1000), B-cell lymphoma 2 (Bcl-2) (#12789-1-AP, 1:2000), Bcl-2-associated X (Bax) (#50599-2-Ig, 1:6000), Nucleoporin p62 (p62) (#18420-1-AP, 1:1000), LC3 (#14600-1-AP, 1:1000), beclin1 (#11306-1-AP, 1:1000), β-actin (#14395-1-AP, 1:15,000) or lamin B1 (#12987-1-AP, 1:1000) (all from Proteintech) and phospho-glucocorticoid receptor (P-GR) (Ser211), extracellular signal–regulated kinases (ERK) (#4161, 1:1000), phospho-ERK (Thr202/Tyr204) (#4377, 1:1000), AMPK (#9158, 1:1000), phospho-AMPK (Thr172) (#5759, 1:1000) were used as internal controls. The membranes were rinsed with TBST five times for 10 min each and incubated with the following secondary antibodies: horseradish peroxidase (HRP)-conjugated Affinipure goat anti-mouse Immunoglobulin G (IgG) (H+L) (SA00001, 1:8000; Proteintech) or HRP-conjugated Affinipure goat anti-rabbit IgG (H+L) (SA00001-1, 1:8000; Proteintech) for 1.5 h at room temperature. Membranes were then rinsed as previously mentioned, and treated with Chemiluminescent HRP Substrate (Millipore, Burlington, MA, USA), which was detected using a chemiluminescence detector (Bio-Rad, Hercules, CA, USA). The expression of each protein was measured using Image Lab software (Bio-Rad).
2.12. Cell Immunofluorescence
HT22 cells were seeded on slides and incubated for 24 h at 37 °C, followed by incubation with 200 μM CORT for 24 h; they were then fixed with 4% paraformaldehyde and seeded on poly-L-lysine-coated coverslips. The cells were permeabilized with 0.3% Triton X-100, blocked with 3% bovine serum albumin, and incubated overnight at 4 °C with antibodies against GR (Proteintech; 24050-1-AP, 3:100), followed by CL488-conjugated Affinipure donkey anti-mouse IgG (H+L) (Proteintech; SA00006-5, 1:200). Nuclei were stained with 4′,6-diamidino-2-phenylindole and the slides were viewed with a laser scanning confocal microscope.
2.13. Transmission Electron Microscopy
After CORT treatment, the cells were washed twice with ice cold PBS, and fixed with 2.5% glutaraldehyde in 0.15 mM sodium cacodylate at 4 °C overnight, then postfixed in 2% osmium tetroxide. All samples were dehydrated in ethanol and embedded in epoxy resin. Then, ultrathin sections (80 nm) of adherent cells were obtained using an ultramicrotome (EM UC7, Leica, Wetzlar, Germany). The sections were counterstained with uranyl acetate and lead citrate and observed using a JEM SX 100 electron microscope (Jeol, Tokyo, Japan) to capture images reference from our previous study [
13].
2.14. Statistical Analysis
Statistical analyses were performed using Prism v.7.0 software (GraphPad Inc., La Jolla, CA, USA). Values are expressed as mean ± SD. Statistical comparisons were performed across different treatment groups (room temperature and cold exposure) and between sexes (male and female) by two-way analysis of variance and in vitro data were analyzed by one-way analysis of variance. p < 0.05 was considered statistically significant.
4. Discussion
The study was focused on the potential mechanism of autophagy and cold exposure in the hippocampal CA1 and CA3 regions of mice, and has shown the relationships of CORT exposure, mitochondrial damage, and autophagy. In our previous results, we found that cold stress was induced following cold exposure, and that the HPA axis, hyperaction-mediated, and CORT excess secretion destroyed the homeostasis of the hippocampus and caused oxidation stress, inflammation, and neuronal apoptosis. Based on these previous results, we hypothesized that cold stress impairs hippocampus function by continually increasing the level of CORT-induced mitochondrial damage, resulting in autophagy. To confirm the hypothesis of autophagy in hippocampus following cold exposure, in vitro, we investigated the potential mechanism between CORT exposure, mitochondrial damage, and autophagy using CORT treatment of HT22 cells. The results showed that HT22 cell autophagy was induced by mitochondrial damage after CORT treatment, and that the mitochondrial damage and autophagy were greater after GR inhibition by RU486.
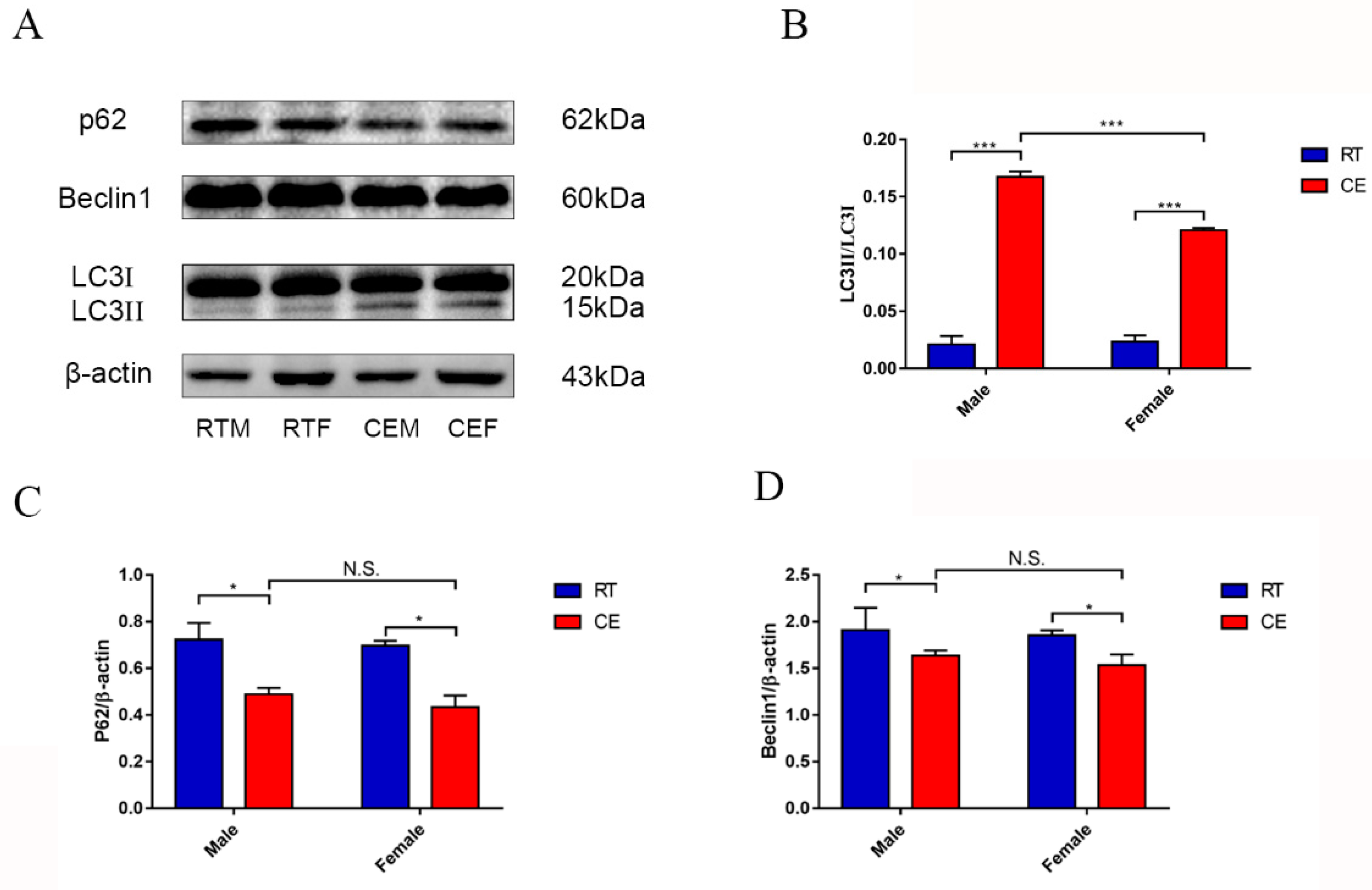
In vivo, the first chronic cold stress model was developed based on our previous report [
8]. As previously mentioned, the CORT level was significantly increased after cold exposure, and oxidation stress occurred in the hippocampus. It is well-accepted that autophagy is the main pathway to remove damaged cell organelles to protect or reconstruct the hippocampus. There have been numerous reports that autophagy occurs in the hippocampus of mice after stress [
15]. In the present report, the results showed that a marker of autophagy, LC3 II, was increased, and the relevant functional protein, p62, was downregulated following cold exposure. The completed autophagosome closure needs LC3 I transformation to LC3 II, because LC3 II is on the vesicle’s inner side and LC3 I was on the outer side. With completion of autophagosomal fusion, the LC3 II is degraded along with the cargo by ubiquitination, and the LC3 I is cleaved and recycled [
16,
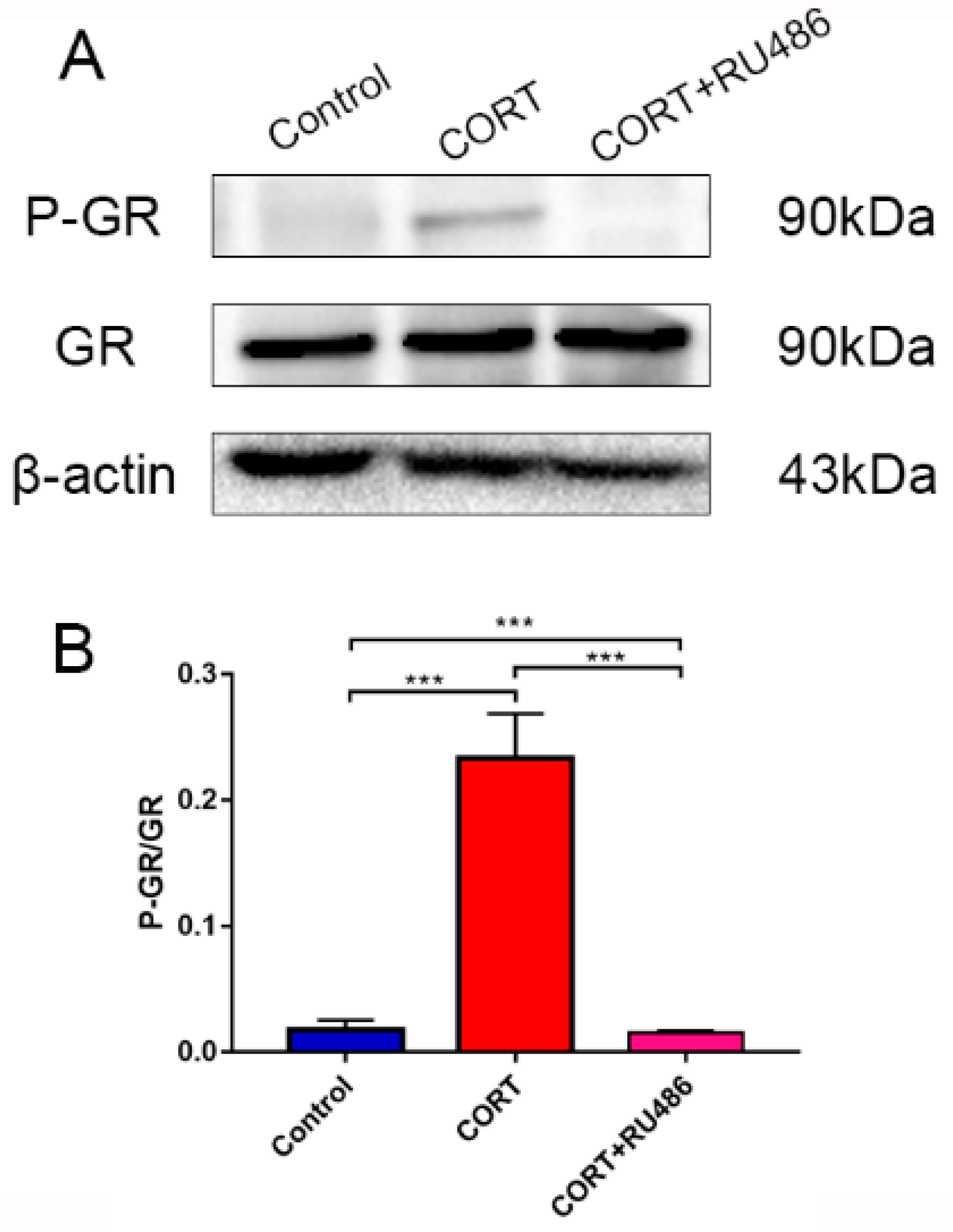
17]. In our results, the phenomenon of autophagy may occur in order to eradicate damaged organelles. To confirm the hypothesis and relevant mechanism, we used 400 μM CORT to treat HT22 cells to develop a CORT exposure model corresponding to the time of cold exposure in vivo, showing that the GR was inhibited by RU486, which confirmed the association between CORT and autophagy. After treatment with CORT, western blots showed that the activation of GR as well as its nuclear translocation were inhibited by RU486.
The apoptosis level after CORT treatment was then measured by FITC-labeled PI and annexin V staining, indicating that the apoptosis level was increased in the CORT-treated group, and that the apoptosis level was even greater in the CORT + RU486 group. These results showed that the effect on HT22 cells from excess CORT was associated with the activation of the GR. Next, the function of mitochondrion was measured. The ATP/ADP/AMP levels were measured in the liquid phase, and the results indicated that the AMP level decreased, and that the ratio of AMP and ATP was also significantly downregulated in a time-dependent manner after CORT treatment. Previous studies have shown that the mitochondria are the energy factories of the cell, and that the AMP/ATP ratio reflects energy production [
18]. A decrease in this ratio may indicate that the mitochondria were affected by CORT treatment, with this decreased trend of AMP/ATP found early in the CORT + RU486 group. The results of ∆Ψm also showed that after CORT treatment, membrane permeability was altered. In the ∆Ψm results, red emission signifies healthy mitochondria, because the healthy mitochondria are polarized, and the JC-1 taken up by such mitochondria forms aggregates. Once the ∆Ψm declines and the membrane permeability changes, the JC-1 will not accumulate in the depolarized mitochondria and is leaked into the cytoplasm, so the monomers will be green [
13]. Based on this staining specificity, the JC-1 staining showed a significant decline in the ∆Ψm of HT22 cells after CORT treatment, indicating that CORT exposure resulted in significant damage to mitochondria, with the membrane permeability changes in RU486 + CORT group being even more severe. The western blotting results also demonstrated the same phenomenon. The ratio of Bcl-2/Bax significantly declined and the expression of Cyt-c increased. As previously reported, Bax forms a heterodimer with Bcl-2, then interacts with the mitochondrial voltage-dependent anion channel with increased openings leading to a loss in membrane potential and the release of cytochrome c, signifying mitochondrial damage [
19,
20]. Here, we confirmed that excess CORT induced mitochondrial damage, and that this damage was more severe when the GR was inhibited. The GR is located in the cytosol under physiological conditions, but it is translocated to the nucleus when it combines with CORT [
21,
22]. The respiration homeostasis of mitochondrial is the most important process in cell survival, and excess CORT may influence the energy cycle and cell respiration. When the GR is inhibited, the receptor is not functional, and excess CORT accumulating in the cytoplasm influences the activation of mitochondria to cause mitochondrial damage. As previously mentioned, autophagy is used to eradicate damaged cell organelles, and western blotting and MDC staining both indicated that the level of autophagy was increased after CORT treatment, which was greater when GR was inhibited. The results of electron microscopy confirmed our hypothesis that the integrity of mitochondria was compromised, and that the damage was more extensive in the CORT and RU486 + CORT group.
Finally, to confirm the hypothesis and potential mechanism of autophagy, the activations of relevant signaling pathways were measured. Western blotting showed that the phosphorylation of AMPK (Thr172) and mTOR (Ser2488) were both upregulated, while the phosphorylation of ERK was downregulated. The activation of AMPK relies on the level of AMP, so the binding of AMP and ADP, and the phosphorylation of AMPK could be induced when the ratio of AMP/ATP was unbalanced [
23]. When AMPK was phosphorylated and activated, the downstream phosphorylation of mTOR was increased and the autophagy process was reversed and activated [
24]. Furthermore, it has been reported that glucocorticoids can affect the kinase activity of MAPK, so excess CORT influenced cell survive by inhibiting phosphorylation of ERK.
In our results, we demonstrated the difference response in hippocampus between male and female mice following cold exposure. Some research has reported there were difference reaction between distinctive gender animals under stress for the reason that the estrogen plays an important role in the process [
25]. In the future, we will still focus on the potential mechanism on the impact between different gender mice following cold stress. Overall, our results indicated that repeated CORT exposure induced mitochondrial damage, which increased neuronal autophagy in the hippocampus during cold exposure (
Figure 12), resulting in an increased risk of neurodegenerative disorders.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











