Neuroprotective Effects of neuroEPO Using an In Vitro Model of Stroke



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Glutamate Exposure
2.3. Cell Death Assays
2.4. Determination of Oxidative Stress
2.5. Statistics
3. Results
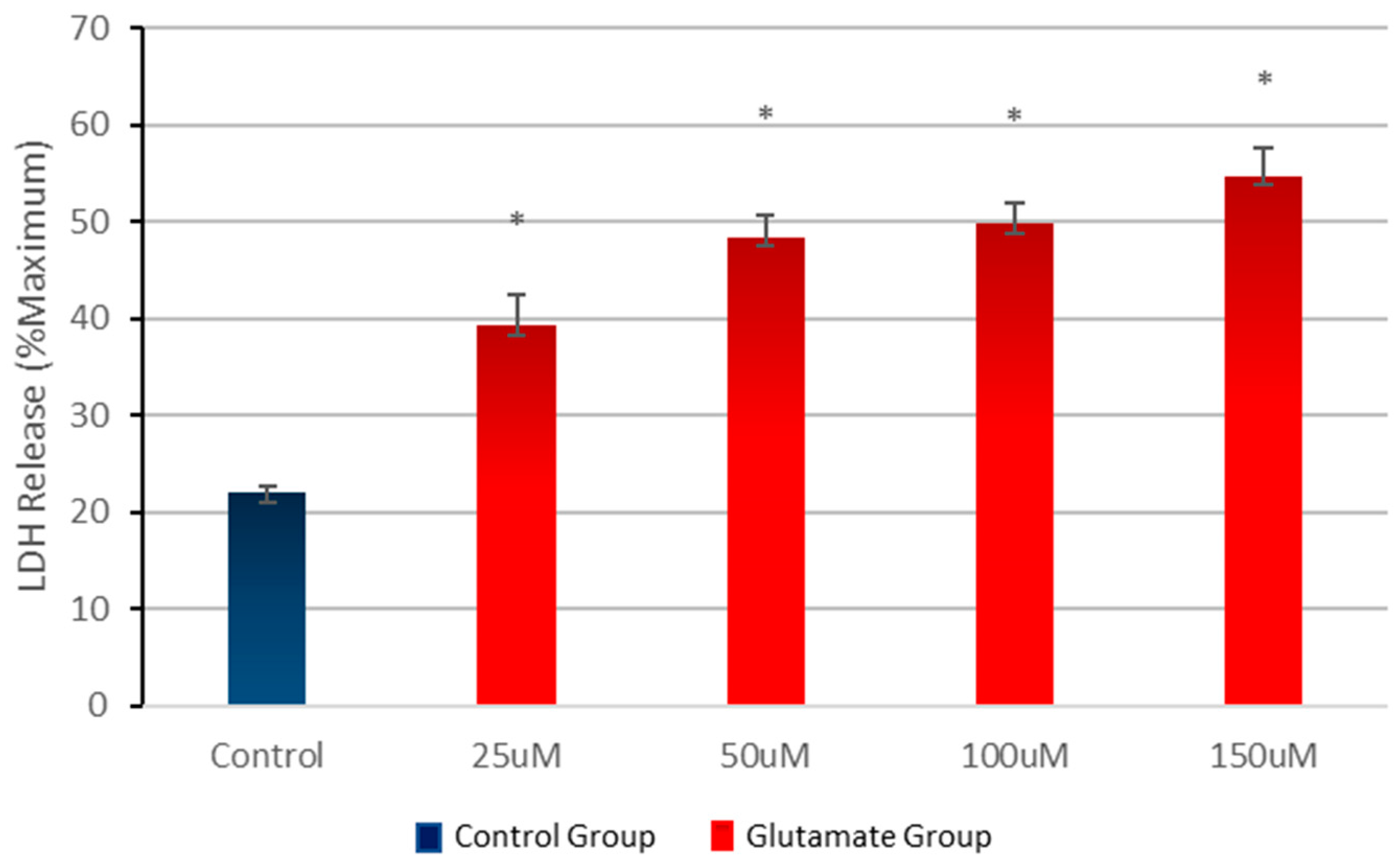
3.1. Effect of Different Concentrations of Glutamate on Cell Viability
3.2. Effect of Different Concentrations of neuroEPO on Cell Viability
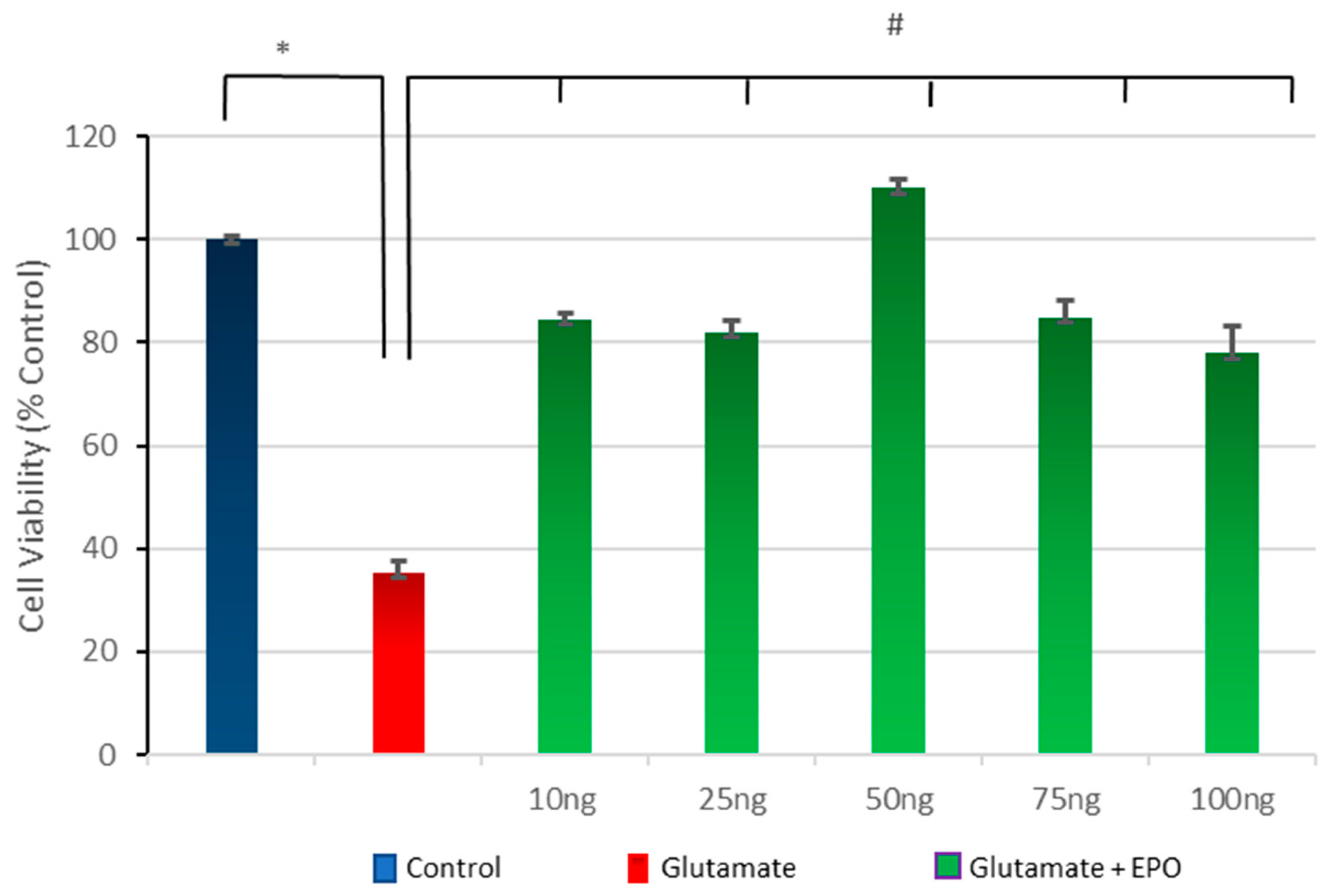
3.3. Effect of Different Concentrations of neuroEPO on Cell Viability after Treatment with Glutamate
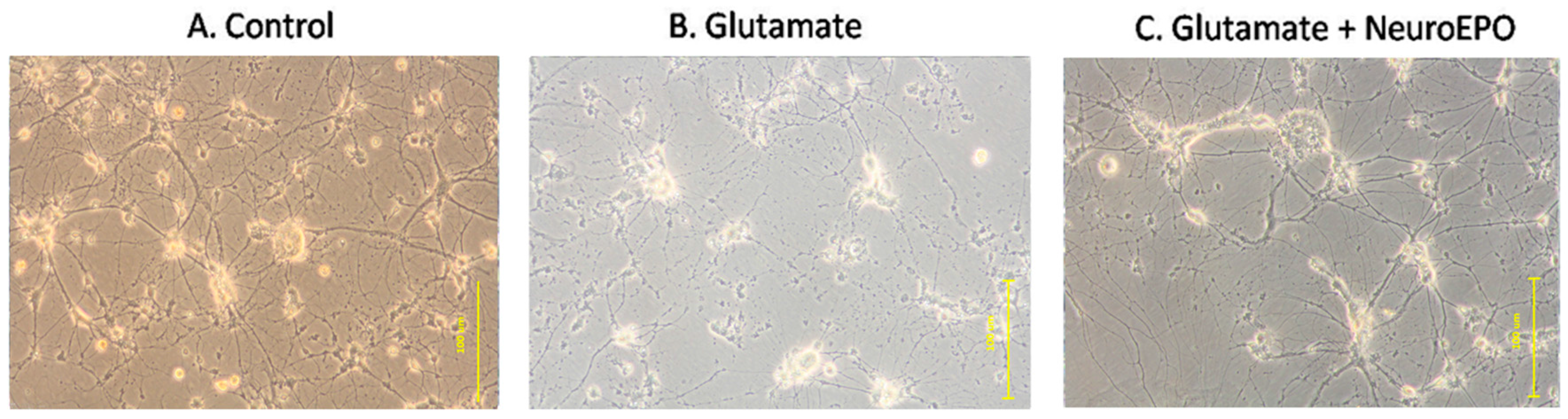
3.4. Effect of neuroEPO on Morphological Changes Induced by Glutamate Excitotoxicity in Neuronal Culture
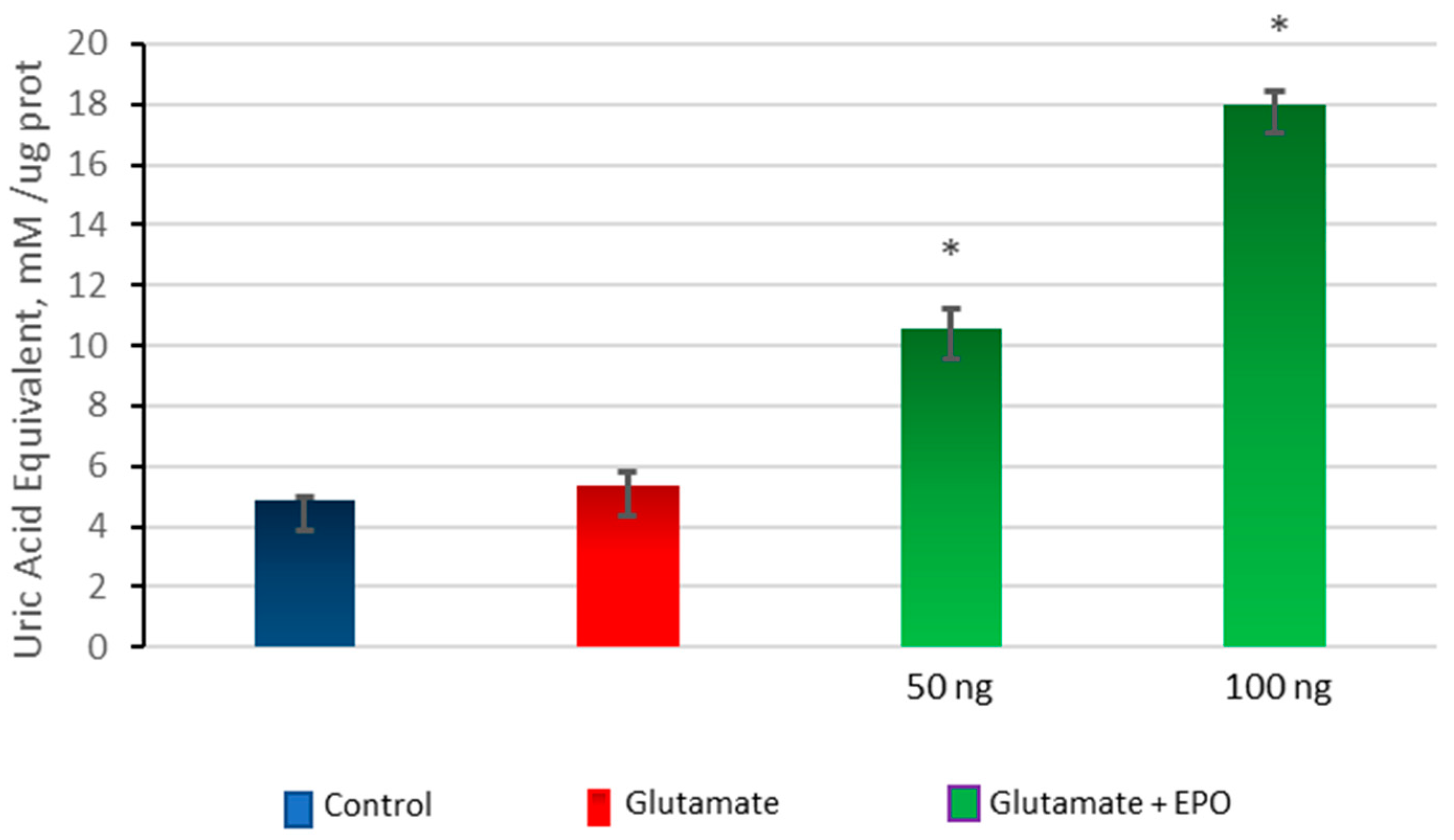
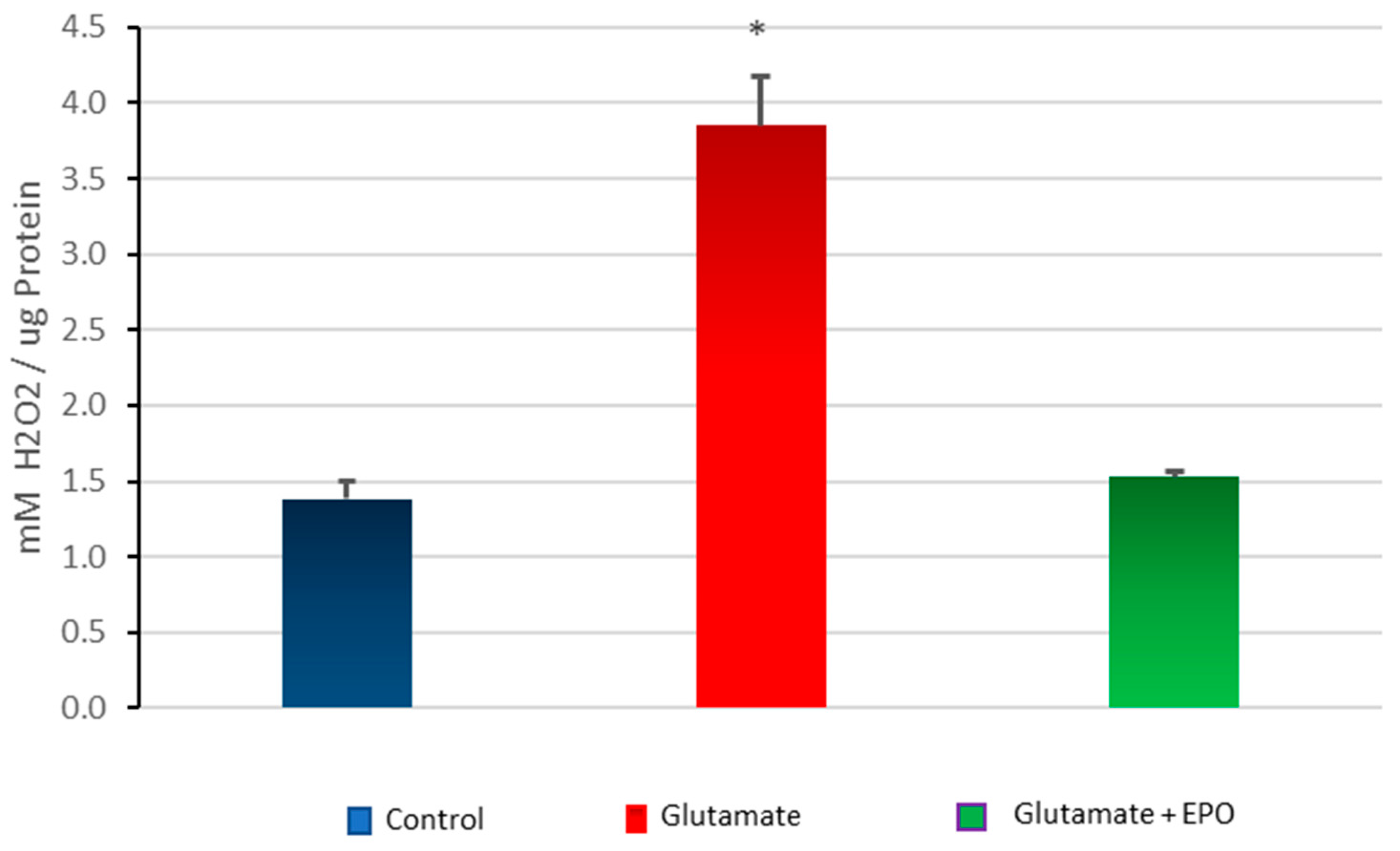
3.5. Effect of neuroEPO on Oxidative Stress in Primary Cortical Cells Treated with Glutamate
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hankey, G. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Bhalla, A.; Wang, Y.; Rudd, A.; Wolfe, C.D.A. Differences in outcome and predictors between ischemic and intracerebral hemorrhage: The South London Stroke Register. Stroke 2013, 44, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Rami, A.; Bechmann, I.; Stehle, J.H. Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog. Neurobiol. 2008, 85, 273–296. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Ferrari, F.; Villa, R.F. Neuroprotection for ischaemic stroke: Current status and challenges. Pharmacol. Ther. 2015, 146, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, T.M.; Thundyil, J.; Tang, S.-C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Steinberg, G.K. Novel stroke therapeutics: Unraveling stroke pathophysiology and its impact on clinical treatments. Neuron 2015, 87, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Erythropoietin after a century of research: Younger than ever. Eur. J. Haematol. 2007, 78, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, M.; Wen, T.C.; Matsuda, S.; Masuda, S.; Morishita, E.; Nagao, M.; Sasaki, R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 1998, 95, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, M.; Marti, H.H.; Roussel, S.; Divoux, D.; Nouvelot, A.; MacKenzie, E.T.; Petit, E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cereb. Blood. Flow Metab. 1999, 19, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Hasselblatt, M.; Dembowski, C.; Cepek, L.; Lewczuk, P.; Stiefel, M.; Rustenbeck, H.-H.; Breiter, N.; Jacob, S.; Knerlich, F.; et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol. Med. 2002, 8, 495–505. [Google Scholar] [PubMed]
- Ehrenreich, H.; Weissenborn, K.; Prange, H.; Schneider, D.; Weimar, C.; Wartenberg, K.; Schellinger, P.D.; Bohn, M.; Becker, H.; Wegrzyn, M.; et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke 2009, 40, e647–e656. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.-K.; Tsai, T.-H.; Lin, H.-S.; Chen, S.-F.; Sun, C.-K.; Leu, S.; Yuen, C.-M.; Tan, T.-Y.; Lan, M.-Y.; Liou, C.-W.; et al. Effect of erythropoietin on level of circulating endothelial progenitor cells and outcome in patients after acute ischemic stroke. Crit. Care 2011, 15, R40. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.Q.; Cherry, B.H.; Scott, G.F.; Ryou, M.-G.; Mallet, R.T. Erythropoietin: Powerful protection of ischemic and post-ischemic brain. Exp. Biol. Med. 2014, 239, 1461–1475. [Google Scholar] [CrossRef] [PubMed]
- Digicaylioglu, M. Erythropoietin in stroke: Quo vadis. Expert Opin. Biol. Ther. 2010, 10, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Jerndal, M.; Forsberg, K.; Sena, E.S.; Macleod, M.R.; O’Collins, V.E.; Linden, T.; Nilsson, M.; Howells, D.W. A systematic review and meta-analysis of erythropoietin in experimental stroke. J. Cereb. Blood Flow Metab. 2010, 30, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Chateauvieux, S.; Grigorakaki, C.; Morceau, F.; Dicato, M.; Diederich, M. Erythropoietin, erythropoiesis and beyond. Biochem. Pharmacol. 2011, 82, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Erbayraktar, S.; Grasso, G.; Sfacteria, A.; Xie, Q.; Coleman, T.; Kreilgaard, M.; Torup, L.; Sager, T.; Erbayraktar, Z.; Gokmen, N.; et al. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 6741–6746. [Google Scholar] [CrossRef] [PubMed]
- Leist, M. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 2004, 305, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Cruz, Y.; Támos Mengana, Y.; Muñoz Cernuda, A.; Subirós Martines, N.; González-Quevedo, A.; Sosa Testé, I.M.; García Rodríguez, J.C. Treatment with nasal neuro-epo improves the neurological, cognitive, and histological state in a gerbil model of focal ischemia. Sci. World J. 2010, 10, 2288–2300. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rodriguez, J.C.; Sosa-Teste, I. The nasal route as a potential pathway for delivery of erythropoietin in the treatment of acute ischemic stroke in humans. Sci. World J. 2009, 9, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Cruz, Y.R.; Strehaiano, M.; Rodríguez Obaya, T.; Rodríguez, J.C.G.; Maurice, T. An intranasal formulation of erythropoietin (Neuro-EPO) prevents memory deficits and amyloid toxicity in the APPSwe transgenic mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 55, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Kussmaul, L.; Hamprecht, B. Detoxification of exogenous hydrogen peroxide and organic hydroperoxides by cultured astroglial cells assessed by microtiter plate assay. Brain Res. Protoc. 1998, 2, 223–228. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Choi, D.W.; Maulucci-Gedde, M.; Kriegstein, A.R. Glutamate neurotoxicity in cortical cell culture. J. Neurosci. 1987, 7, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.S.; Pascoe, C.J.; Giardina, S.F.; John, C.A.; Beart, M.P. Micromolar l-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacol. 1998, 37, 1419–1429. [Google Scholar] [CrossRef]
- Vaarmann, A.; Kovac, S.; Holmström, K.M.; Gandhi, S.; Abramov, A.Y. Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis. 2013, 4, e455. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Greenwood, S.M.; Connolly, C.N. Dendritic and mitochondrial changes during glutamate excitotoxicity. Neuropharmacology 2007, 53, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Castilho, R.F.; Ward, M.W.; Nicholls, D.G. Oxidative stress, mitochondrial function, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem. 1999, 72, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Hayashi, A.; Sun, H.-S.; Belmares, M.P.; Cobey, C.; Phan, T.; Schweizer, J.; Salter, M.W.; Wang, Y.T.; Tasker, R.A.; et al. PDZ protein interactions underlying nmda receptor-mediated excitotoxicity and neuroprotection by PSD-95 inhibitors. J. Neurosci. 2007, 27, 9901–9915. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef]
- Szydlowska, K.; Tymianski, M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010, 47, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Stanika, R.I.; Pivovarova, N.B.; Brantner, C.A.; Watts, C.A.; Winters, C.A.; Andrews, S.B. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9854–9859. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Kleikers, P.W.M.; Wingler, K.; Hermans, J.J.R.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H.H.W. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Andoh, T.; Echigo, N.; Kamiya, Y.; Hayashi, M.; Kudoh, I.; Goto, T. Effects of erythropoietin on intracellular calcium concentration of rat primary cortical neurons. Brain Res. 2011, 1387, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Tymianski, M. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 2000, 78, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Tymianski, M. Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat. Neurosci. 2011, 14, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- Morishita, E.; Masuda, S.; Nagao, M.; Yasuda, Y.; Sasaki, R. Erythropoetin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 1996, 76, 105–116. [Google Scholar] [CrossRef]
- Calapai, G.; Marciano, M.C.; Corica, F.; Allegra, A.; Parisi, A.; Frisina, N.; Caputi, A.P.; Buemi, M. Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur. J. Pharmacol. 2000, 401, 349–356. [Google Scholar] [CrossRef]
- Sinor, A.D.; Greenberg, D.A. Erythropoietin protects cultured cortical neurons, but not astroglia, from hypoxia and AMPA toxicity. Neurosci. Lett. 2000, 290, 213–215. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernando, G.; Yamila, R.; Cesar, G.J.; Ramón, R. Neuroprotective Effects of neuroEPO Using an In Vitro Model of Stroke. Behav. Sci. 2018, 8, 26. https://doi.org/10.3390/bs8020026
Fernando G, Yamila R, Cesar GJ, Ramón R. Neuroprotective Effects of neuroEPO Using an In Vitro Model of Stroke. Behavioral Sciences. 2018; 8(2):26. https://doi.org/10.3390/bs8020026
Chicago/Turabian StyleFernando, Garzón, Rodríguez Yamila, García Julio Cesar, and Rama Ramón. 2018. "Neuroprotective Effects of neuroEPO Using an In Vitro Model of Stroke" Behavioral Sciences 8, no. 2: 26. https://doi.org/10.3390/bs8020026
APA StyleFernando, G., Yamila, R., Cesar, G. J., & Ramón, R. (2018). Neuroprotective Effects of neuroEPO Using an In Vitro Model of Stroke. Behavioral Sciences, 8(2), 26. https://doi.org/10.3390/bs8020026