1. Introduction
Marine biorefineries, based around the valorization of salt water macroalgae, have been suggested as a promising improvement to terrestrial alternatives [
1,
2]. Macroalgae are photosynthetically efficient, do not compete with agricultural land, do not contain lignin and represent a largely untapped bioresource. To this end, a large body of research has been invested in the valorization of macroalgae, including pretreatments, fermentation and thermochemical conversion routes [
3,
4,
5]. A considerable number of challenges remain however, including having the ability to convert multiple species with highly variable composition [
6,
7], to handle salt water as part of the process [
8] and to be able to cope with either heavy metal [
9] or plastic contamination [
10]. As such, it seems likely that feedstock agnostic processes, which can handle a wide variability and produce an array of products, will be critical to further development in this field. One such processing methodology is hydrothermal liquefaction (HTL), a promising thermochemical pathway identified as a cost-competitive process for converting high-moisture biomass. HTL avoids energy losses associated with drying, which is needed for other thermochemical processes [
11], delivers a high-energy biocrude (30–40 MJ kg
−1) [
11] and produces a more stable crude product than pyrolysis [
12,
13,
14]. HTL generally uses temperatures between 280 and 370 °C and pressures between 10 and 25 MPa to maintain water in the liquid state [
15]. Water acts as an important reactant and the addition of catalyst leads to several opportunities for separations and further chemical reactions [
16,
17]. The reactions that take place during HTL are decomposition and repolymerization to form biocrudes with high heating values, a solid residue containing the inorganic fraction and a water-soluble fraction that can be used as a fertilizer [
18].
Contamination of oceans by microplastic and macroplastic debris has become one of the most publicized marine environmental issues of recent years, affecting all of the world’s oceans. For instance, microplastics have been demonstrated to affect the growth of macroalgae. Alarmingly, ingestion of plastic debris has been recorded in 44–50% of all seabirds. This leads to blocking of the digestive tract, damage stomach lining, less feeding and starvation [
19]. Plastics have been accumulating in substantial densities in the marine environment, from the sea surface down to deep-sea sediments [
20,
21]. In a marine biorefinery, macroalgae could possibly be washed in clean water to remove microplastics, but residual microplastics can be absorbed onto the surface of marine macroalgae, or be embedded in the structure [
22]. Macroplastic, on the other hand, would need to be removed manually. Marine plastics originate mainly from many types of plastic debris, such as fishing nets, ropes and plastic bags. While most of the focus has been on terrestrial PET and polyolefin litter, over 20% of marine plastic debris found in the ocean is estimated to come from commercial fishing activity [
23]. For example, an estimated 640,000 tonnes of nylon fishing gear enters the oceans every year, which amounts to approximately 10% of the total marine debris [
24]. These discarded fishing items, including monofilament lines and nylon netting used in fishing activity, have contributed to the considerable growth in the marine plastic contamination that is having global impact on the entanglement of marine life [
23]. Furthermore, due to the nature of the fibers, nylon fishing lines are one of the main contaminants found in macroalgal beds and seaweed farms [
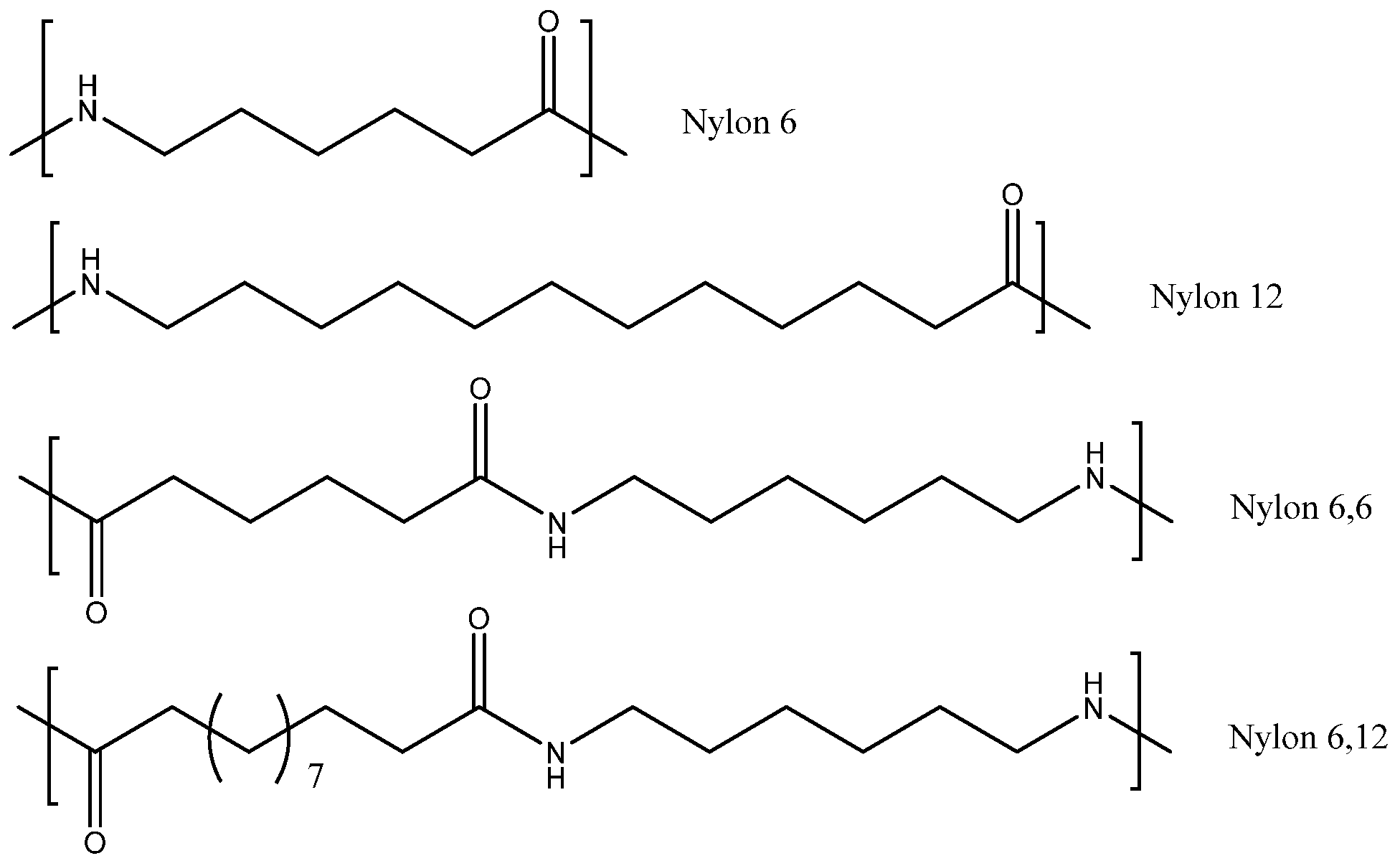
10]. Nylon refers to simple polyamides (
Figure 1). For example, nylon 6 is synthesized by the ring opening polymerization of caprolactam [
25]. While the majority of maritime fishing lines are produced from nylon 6, there is increasing concern that alternative nylon polymers such as nylon 6/6, nylon 12 and nylon 6/12 are increasingly distributing into the oceans from synthetic textile fibers that have been worn down during washing.
Research on hydrothermal liquefaction of marine macroalgae has gained significant traction in recent years [
26,
27,
28,
29,
30], and a number of publications have investigated the co-liquefaction of marine biomass with plastics. Wu et al. reported the co-liquefaction of microalgae with polypropylene and found that the composition of the biocrude products was improved with reducing acid content [
31]. A similar observation was made by Coma et al. [
32], for the co-liquefaction of Spirulina and
Ulva sp. with polyolefins distributing partially into the crude phase. In a more in depth study, Hongthong et al. demonstrated that a range of plastics could be co-processed with pistachio hulls as biomass through hydrothermal liquefaction, including nylon 6, which broke down almost completely (>80%) under HTL conditions [
33]. Similarly, Raikova et al. screened multiple plastics, including nylon 6 in the co-liquefaction of macroalgae. In this study, they demonstrated that nylon 6 did breakdown under HTL conditions and one of the monomers, ϵ-caprolactam was observed, predominantly in the aqueous phase [
34].
Plastics are one of the most promising resources for fuel production, with an average high heating value of 38–46 MJ/kg [
35], compared to 14–16 MJ/kg for macroalgae biomass [
29]. Moreover, the high content of metal in the ash of macroalgae biomass can play an important catalytic role in thermochemical processes [
36,
37]; as a result, coprocessing may lead to synergistic effects that promote the decomposition of biomass as well as plastic waste [
38]. Therefore, the conversion of macroalgal biomass and marine plastic could not only reduce costs through no longer needing to separate the two components, but could also lead to a significant enhancement in the efficiency and economics of a process, as well as provide environmental protection. Moreover, with evidence that monomers of nylon 6 could be selectively produced [
34], this could significantly add to the value of a marine biorefinery by enabling the chemical recycling of waste nylon alongside the production of biocrude and fertilizer products.
This work builds on the promising results for nylon 6, and systematically assesses the hydrothermal liquefaction of macroalgae biomass with a range of nylons commonly found in maritime plastic waste, including nylon 6, nylon 6/6, nylon 6/12 and nylon 12. The technique was also applied to an actual sample of marine macroalgae collected at sea, entangled with nylon fishing line to demonstrate the concept.
2. Materials and Methods
2.1. Material
Fresh
F. serratus samples were collected from Saltern Cove, Paignton, Devon, UK. Prior to analysis, all samples were freeze-dried and milled to 10 μm diameter. Nylon 6, nylon 6/6, nylon 6/12 and nylon 12 were obtained from Sigma-Aldrich (Dorset, UK) and used without further purification. All samples were stored at ambient conditions. The original composition (C, H and N) of the raw material was measured by elemental analysis and carried out externally at London Metropolitan University (London, UK), on a Carlo Erba Flash 2000 Elemental Analyzer. The analysis is shown in
Supplementary Material (Table S1).
2.2. Hydrothermal of Coliquefaction of Nylon and Microalgae
Hydrothermal liquefaction of nylon and
F. serratus was performed in a stainless-steel (Swagelok Tube Fitting, Bristol UK) batch reactor according to a previous report [
34]. The reactor had an approximate internal volume of 50 mL, and was connected with a pressure gauge, thermocouple, needle valve and relief valve. The temperature was monitored using a thermocouple connected to data logging software (K-type thermocouple, RS component, Leeds, UK, data acquisition with an ADAM-4018+data logger, and collected with software written in LabVIEW 2011). The reactor was not pressurized and loaded with a total of 3 g biomass (
F. serratus mixed with nylon: 100:0, 95:5, 80:20 and 50:50) and 15 g of distilled water and heated within a vertical tubular furnace (Carbolite EVA 12/300B, Essex, UK) until the temperature reached 350 °C. The reactor was then removed from the furnace and allowed to cool to room temperature. Total heating time was approximately 10 min with an average heating rate of 35 °C min
−1. The experiments were repeated three times under the same conditions.
2.3. Calculation of HTL Products
After cooling, gaseous products were released via the needle valve into an inverted, water-filled measuring cylinder to measure gaseous fraction volume. The yields of each product phase were calculated as mass percentage on ash-free basis. Gas phase yields were calculated using the ideal gas law, approximating the gas phase as 100% CO
2, assuming an approximate molecular weight of 44 g mol
−1 and a volume of 22.465 dm
3 mol
−1 gas phase at 25 °C. The gaseous product yield was determined using the following equation:
The aqueous phase was separated to eliminate undissolved materials, the reactor contents were filtered by 1 μm pore size filter paper predried overnight at 60 °C. Product yield in the water phase was determined by leaving a 2.0 g aliquot and drying in a 60 °C oven overnight and the aqueous residue yield was calculated by scaling aliquot to the total mass of aqueous phase. Aqueous phase residue yield was determined by using the following equation:
To separate the remaining biocrude and solid residue phase, the reactor was washed repeatedly with chloroform until the solvent was clear, the solution was filtered, and any residual bio-oil washed off the filter paper. The chloroform was removed using a rotary evaporator at 40 °C for 1.5 h to give dark oil (biocrude). Biocrude yield was determined using the following equation:
The solid residue on the filter paper was oven-dried overnight at 60 °C to determine the solid residue product yield. Solid residue biochar yield was determined using the following equation:
Mass loss occurred during the separation process, mainly through the loss of volatile organic compounds during the evaporation of solvent during removal of the biocrude phases through filtration. The overall mass balance of the reaction was calculated according to the following equation:
The nylon conversion in the system was calculated according to our previously published method [
33]. Briefly, known amounts of nylon were added to biochar produced from the HTL of macroalgae; these samples were assessed by FTIR and a calibration curve was built and used to estimate the conversion of the resulting coliquified samples. For full details, see the
Supplementary Materials.
2.4. Characterisation
The moisture content (%) was determined by drying 2 g of
F. serratus. The sample was put into an oven at 105 °C and heated overnight. The dried sample was allowed to cool and then reweighed.
F. serratus ash was measured by heating a 100 mg of
F. serratus in a Carbolite CWF 11 muffle furnace (Carbolite Gero Ltd., Hope Vallage, UK) at 550 °C for 24 h. The mass remaining at the end of the experiment was calculated as the ash content.
F. serratus protein content was calculated from biomass elemental N concentration, with protein mass fraction quoted on dry basis using a conversion factor of 6.25. [
39]
F. serratus total lipid content was extracted from the samples with chloroform/methanol (2:1,
v/
v). Approximately 200 mg of dried
F. serratus was placed into a tube, 14 mL of the chloroform–methanol mixture added and after 2 min in a vortex mixer the contents of the tube were filtered through 1 μm pore size filter paper. The filtered extract was washed with a chloroform–methanol mixture. The chloroform–mixture was removed using a rotary evaporator. The solid retained was determined as the total content of the macroalgae biomass.
Total carbohydrate was calculated, determined by difference:
where
X_component is the mass fraction (%) of each biochemical component.
Elemental analysis (carbon, hydrogen and nitrogen content) of the biomass feedstock and products was carried out externally at London Metropolitan University on a Carlo Erba Flash 2000 Elemental Analyzer. The oxygen content was determined by the difference from the sum of carbon, nitrogen and hydrogen, assuming negligible sulfur in the products.
The higher heating values (
HHV) of the biomass, biochar and biocrude were calculated using data from the elemental composition in the following equation proposed by the Dulong formula [
38], where
C,
H and
N are the weight percentages of each element:
The chemical energy recovery (ER) was calculated for the biocrude and solid residue biochar as follows:
The extent of interactive synergistic effects between the
F. serratus and plastics were calculated in terms of biocrude yield. Theoretical results are determined by comparing the experimental biocrude obtained during cohydrothermal liquefaction with the individual feedstock by the equation below:
where
YB is the yield of biocrude obtained in the HTL experiment,
XM is the mass fraction of
F. serratus in the total reaction mixture,
YM is the biocrude yield of
F. serratus and
YNY is the biocrude yield of pure nylon plastic.
The chemical composition of the volatile fraction of the biocrude was investigated using an Agilent Technologies 8890A GC system (Santa Clara, CA, USA) fitted with a 30 m × 250 µm × 0.25 µm HP5-MS column (Santa Clara, CA, USA), coupled to a 5977B inert MSD (Santa Clara, CA, USA). Samples were dissolved in tetrahydrofuran (THF, Fisher chemical, Loughborough, UK), the carrier gas was helium, with a flow rate 1.2 mL min−1. An initial oven temperature was set to 50 °C, increasing to 250 °C at 10 °C min−1. Initial identification of compounds was performed by means of the mass spectral database in the National Institute of Standards and Technology (NIST) library.
The aqueous phase from HTL was characterized through liquid chromatography using an Acquity UPLC BEH C18, 1.7 μM, 2.1 × 50 mm reverse phase column (Waters, Milford, MA, USA) with a flow rate of 0.3 mL min−1 at 45 °C, and an injection volume of 10 µL. Mobile phases A and B consisted of 0.1% v/v formic acid in water, and 0.1% v/v formic acid in methanol, respectively. Gradient elution was carried out with 5% mobile phase B for 2 min followed by a linear gradient to 100% B for 5 min; these conditions were held at 100% B for 8 min and then returned to 5% B in 12 min.
Total metal content for solid residue and biocrude products were analyzed using an Agilent 7700 Series ICP-MS. Samples were digested in aqua regia. Briefly, 50 mg solid residue was dissolved in 3 g hydrochloric acid (37%; Fisher Tracemetal grade, Fisher chemical, Loughborough, UK); 1 mL concentrated nitric acid (67%; Fisher Tracemetal grade, Fisher chemical, Loughborough, UK) was then added and left to digest at room temperature for 15 min. The digest was subsequently heated to 95 °C for 60 min, and left to digest at ambient temperature overnight and made up to 25 mL with distilled water. The resulting solution was filtered through a 1 µm filter membrane prior to analysis.
3. Results and Discussion
3.1. Product Yields and Distribution
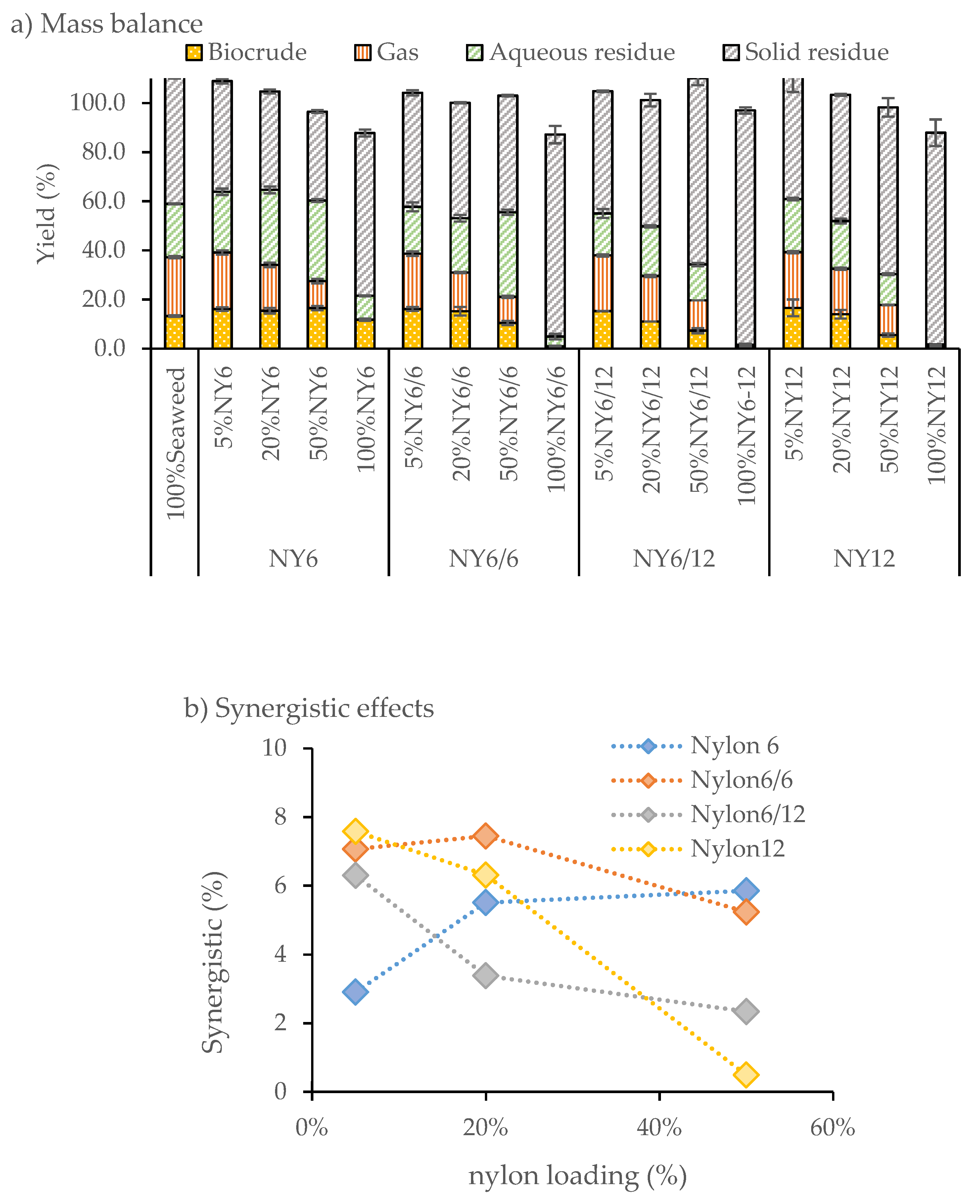
F. serratus was co-processed with 5, 20 and 50 wt.% weight loadings of nylon 6, nylon 6/6, nylon 6/12 and nylon 12 at 350 °C, and a residence time of 10 min. Mass balances where product yields were calculated on the ash-free basis (DAF%) of total feedstock input are shown in
Figure 2. The solid residue char formation can be reduced for co-liquefaction of
F. serratus with nylon 6, where the yield decreased from 52.4% for pure seaweed to 45.1%, 40.1% and 36.2% for 5, 20 and 50 wt.% nylon 6 blends, respectively. Under the same conditions, without
F. serratus, 66.3 wt.% of nylon 6 remained unreacted and distributed into the char phase.
Blending of F. serratus with nylon 6 increased biocrude yields from 13.2% for pure F. serratus to 16.5% for the 50 wt.% nylon 6 blend. On addition of the nylon 6 blend, the gas phase product yield decreased slightly, and no gas was observed in the liquefaction of nylon 6 without F. serratus. The aqueous phase residue yield increased from 21.8% for pure F. serratus to 24.7%, 30.6%, 32.8% for 5, 20, and 50 wt.% nylon 6 blend, respectively. This trend in yield is highly suggestive that the nylon 6 breaks down in the reaction and distributes to both the crude and aqueous phases.
In contrast, the other nylon polymers (nylon 6/6; nylon 6/12 and nylon 12) did not behave in a similar manner under the HTL conditions tested. For the co-liquefaction of nylon 6/6, the solid residue increased on increasing nylon blend wt.% whilst increasing nylon 6/6 blend levels caused a decrease in the yield of biocrude products. This result suggests that limited reactivity and thus breakdown/conversion is occurring. Similarly, little biocrude or aqueous phase residue was recovered from the liquefaction without F. serratus, with the majority of mass retained in the solid residue.
A similar limited reactivity was observed for both nylon 6/12 and nylon 12, where most of the mass was retained in the solid residue. Similar to nylon 6/6, both nylon 6/12 and nylon 12 did not break down significantly in the reaction without F. serratus.
While no nylons readily biodegrade in the natural environment, nylon 6/6 is known to be more thermally stable than nylon 6. Kinetic studies on the thermal decomposition of nylons showed that the minimum activation energy for the decomposition of nylon 6 and nylon 6/6 was 180 kJmol
−1 [
40] and 223 kJmol
−1 [
41], respectively. Activation energy is affected by the process of bond breaking at the C-N bonds, which is the rate-determining step of decomposition in nylon. Nylon 12, on the other hand, has a higher hardness and tensile strength than nylon 6 and nylon 6,6. Nylon 12 contains a lower content of amide groups per unit area, also leading to a lower moisture absorption [
42]. The lower amide group content and increased olefin-type structure results in nylon 12 having a higher resistance to chemical degradation, and higher tensile strength and stiffness. This suggests that nylon 12 and nylon 6/12 are more stable compared to other nylons, and therefore more resistant to being broken down.
There appears to be an interaction between the F. serratus biomass and the nylon blends where the F. serratus synergistically aids the breakdown of nylon plastic. Results revealed that co-processing reduced solid residue biochar formation, and biocrude production was increased during co-liquefaction compared to the individual components.
A positive synergistic effect on biocrude production was observed between nylon 6 and F. serratus on co-liquefaction. This involves the extensive hydrolysis of nylon 6 that yielded ϵ-caprolactam. However, the synergistic effect decreases substantially on the coprocessing of the macroalgae and nylon 6/6, and is negligible on the coprocessing of nylon 6/12 and nylon 12 with the macroalgae. This is consistent with the increasing solid residue observed with these latter reactions.
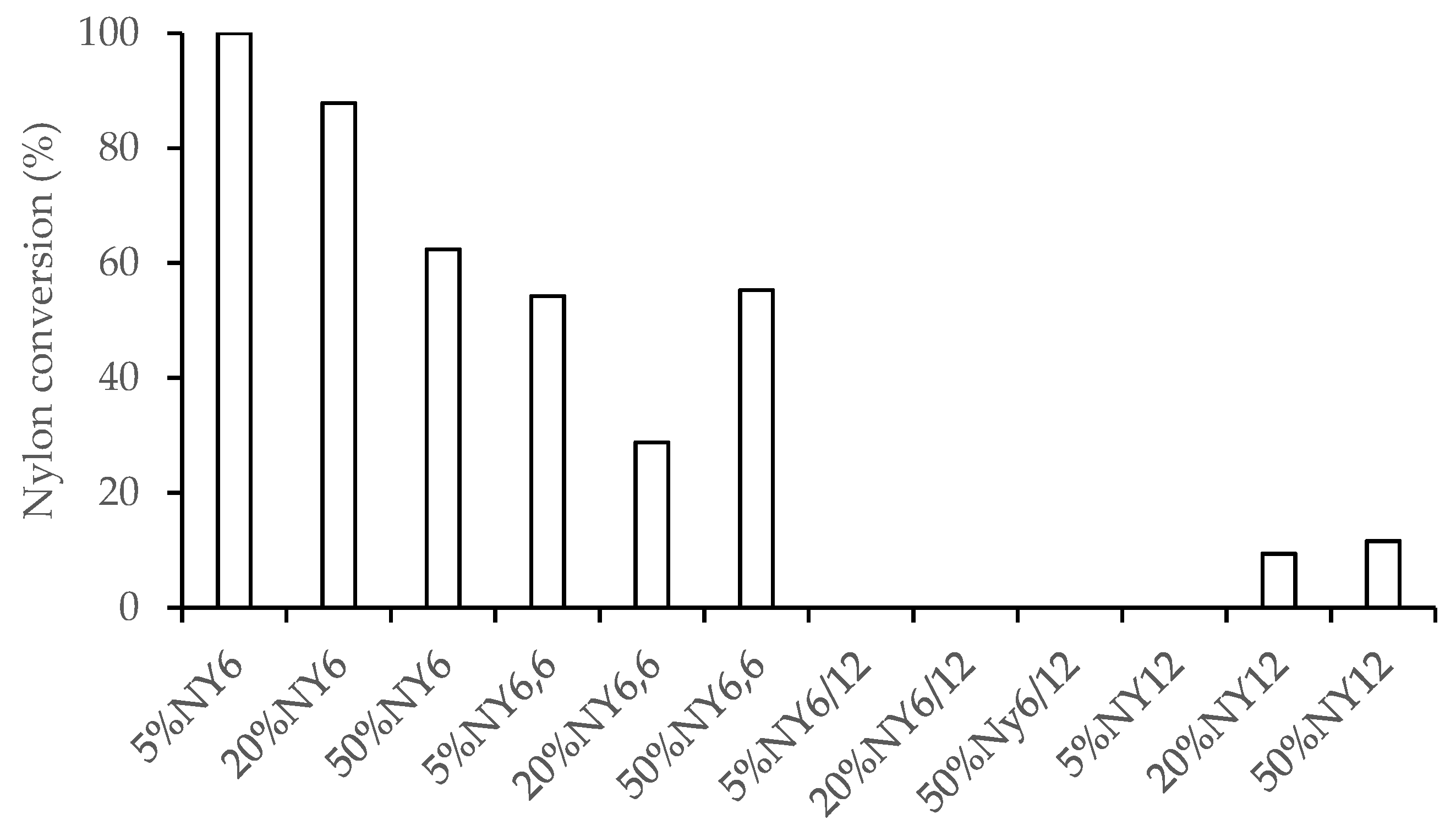
3.2. Nylon Conversion
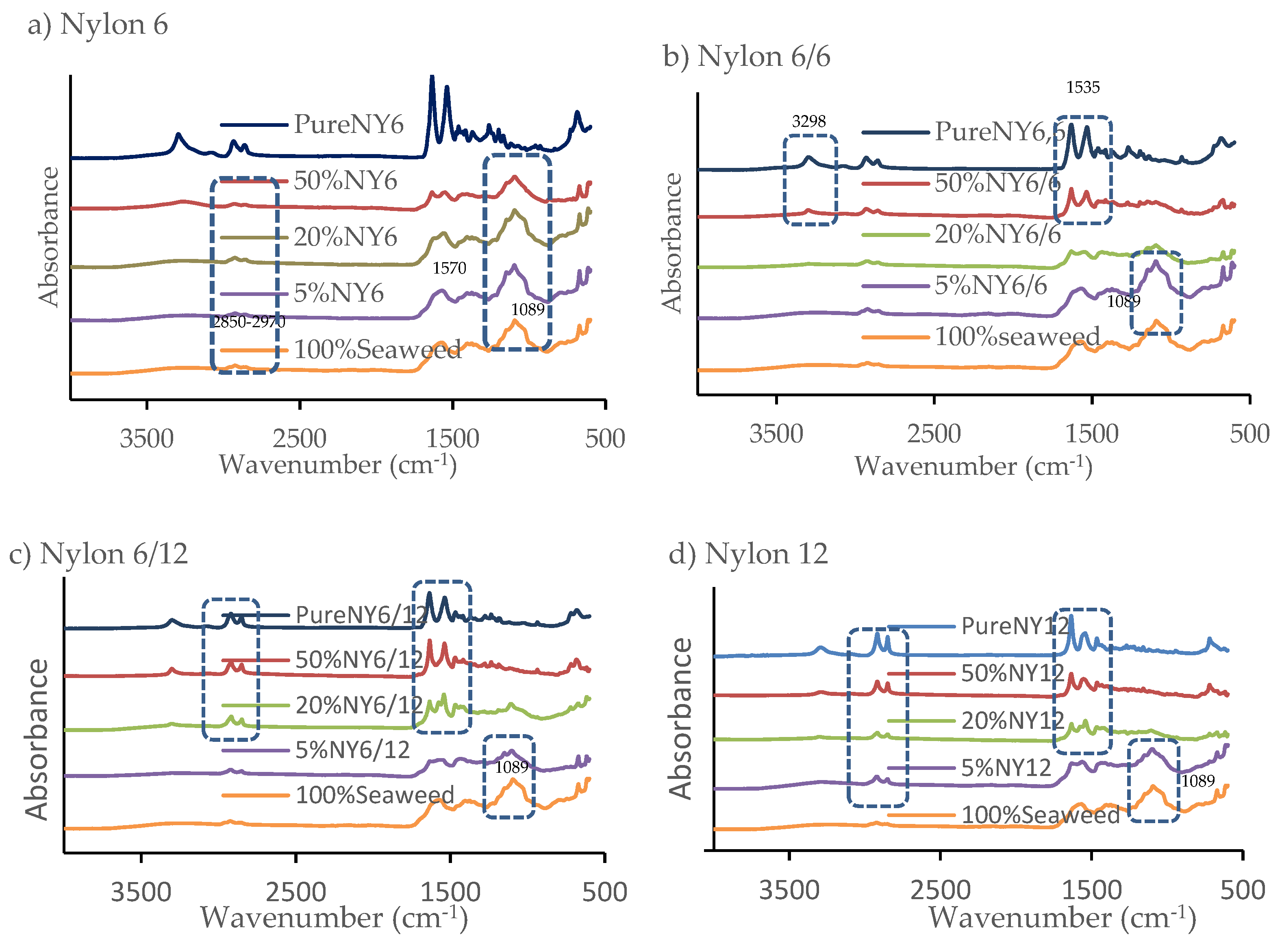
Fourier transform infrared (FTIR) analysis was performed to determine the amount of nylon remaining in the solid residue and therefore estimate the overall nylon conversion (
Figure 3). The FTIR spectra were compared against a standard curve created with increasing known amounts of nylon added to the solid residue obtained from the HTL of pure
F. serratus to estimate the overall conversion (
Figure 4). For the solid residue from co-liquefaction of
F. serratus and nylon 6, peaks of moderate intensity were observed similar to those obtained for solid residue from HTL of pure
F. serratus biomass (peak absorbance at 1089, 1570 and 2850–2970 cm
−1). The presence of nylon 6 co-feedstock in HTL reactions did not appear to change the composition of the biochar. This result suggests that during the co-liquefaction of
F. serratus and nylon 6, there is a synergistic interaction between the two components resulting in an improved decomposition. For example, even with 50 wt.% blends, there are only trace amounts of nylon left in the solid fraction (
Figure 3).
However, on comparing spectra of the biochar from co-liquefaction of
F. serratus with nylon 6/6, the spectra blend closely resembles the solid residue from pure
F. serratus with the addition of 5 wt.% nylon 6/6 blend. The intensity peak at 1089 cm
−1 decreased with increasing the addition of 20 wt.% nylon 6/6, suggesting that the solid residue derived from the addition of 20 wt.% nylon 6/6 had lower ash content than the addition of 5 wt.% nylon 6/6. Peak intensities from HTL of pure
F. serratus bands decreased in almost all the solid residues obtained with the addition of 50 wt.% nylon 6/6 blend, strong peaks were observed at 1535 cm
−1, corresponding to the stretching vibration of N–B–N and N=Q=N where –B– and =Q= stand for benzenoid and quinoid moieties in the polymer [
43]. In addition, a peak was observed at 1631 cm
−1, corresponding to carbonyl groups of nylon 6/6 [
43]. Another relevant band observed at 2949 and 2823 cm
−1 is attributed to CH
2 stretching, and peaks were observed at 3298 cm
−1 corresponding to N–H stretch. These peaks were also observed in the FTIR spectra of pure nylon 6/6, but not seen in the spectra of the biochar from
F. serratus. These findings indicate the presence of a large unreacted portion of nylon 6/6 in the HTL solid residue.
Similar patterns in IR absorbance of the solid residue were observed for nylon 6/12 and nylon 12, where increasing blend levels gave rise to similar increases in the peaks associated with nylon 6/12 and nylon 12. Thus, under the conditions tested only nylon 6 underwent conversion whilst the other nylon species were too stable for conversion.
3.3. Biocrude Composition
The biocrude samples were analyzed by GC–MS (Agilent Technologies 8890A GC system, Santa Clara, CA, USA) to determine the chemical components of the biocrude. A change in the chemical compounds found in the biocrude was observed when adding a range of different nylon blends. The
F. serratus alone produced a biocrude product that contained mainly phenolic compounds potentially derived from the reaction of organic acids and carbohydrates in the system. Under HTL conditions, co-liquefaction with nylon 6 produced an oil phase that contained ϵ-caprolactam, the monomer used in the polymerization of nylon 6. When increasing the nylon 6 at 5, 20, 50 and 100 wt.% blend levels, the level of ϵ-caprolactam increased substantially. This observation suggests that nylon 6 was successfully decomposed by hydrolysis at subcritical conditions to form high yields of ϵ-caprolactam via an ϵ-aminocaproic acid intermediate in biocrude production [
44]. In addition, low levels of ϵ-caprolactam were also present in biocrude from pure
F. serratus, most likely arising from protein decomposition in the marine macroalgae biomass.
During HTL processing, the presence of nylon 6/6 led to an increased formation of cyclopentanone, which is formed by a cyclic degradation mechanism in adipic acid [
45]. High concentrations of cyclopentanone were detected in the presence of 50 wt.% nylon 6/6 blend. In addition, for co-liquefaction of nylon 6/6 blend levels, low levels of ϵ-caprolactam were observed, although these are presumably produced from the original
F. serratus.For the co-liquefaction of nylon 6/12 and
F. serratus, cyclododecanone was observed with the addition of 20 wt.% nylon 6/12. Cyclododecanone a cyclic ketone, an important precursor involved in the synthesis of nylon 6/12 and nylon 12 [
46] and most likely the breakdown product produced from the degradation of the polymer in the system. Both cyclopentanone and cyclododecanone were not observed in the biocrude derived from
F. serratus alone.
Nylon 12 is more stable than nylon 6, partly as the polymer contains a lower concentration of amide groups [
47]. A small amount of cyclododecanone was obtained following the conversion, which originates from nylon 12. However, the main chemical components are similar to biocrude observed from pure macroalgae, containing mainly phenol and 2-Cyclopenten-1-one, 2-methyl.
The elemental composition of the biocrude products obtained from co-liquefaction of
F. serratus and nylon blends is presented in
Figure 5. Hydrothermal liquefaction of pure
F. serratus alone included a substantial change in the elemental composition compared to the pure unprocessed
F. serratus feedstock, resulting in a substantial carbon increase and oxygen decrease (C increasing from 35.2% to 71.3% and O decreasing from 57.5% to 15.9%). With an increasing nylon blend level, the overall impact on biocrude element composition was similar to that observed for
F. serratus alone; however, different trends were found in the presence of nylon 6 blends. Carbon content decreased slightly with the increase of nylon 6/6, nylon 6/12 and nylon 12 blend levels, but decreased substantially for the nylon 6 blend (which gave a decrease from 71.3% for pure macroalgae to 60.4% for 50 wt.% nylon 6 blend). There are no significant differences in elemental hydrogen and nitrogen composition in the biocrude samples obtained from the presence of nylon blends (ranging from 8.4 to 9.5 for hydrogen content and between 4.3 and 7.9 for nitrogen content). A slight increase in both hydrogen and nitrogen was observed with increasing nylon content in all blends. This suggests that
F. serratus biomass and nylon radicals may contribute through a biomolecular termination reaction, where a hydrogen radical is transferred from a nylon chain to biomass radicals [
48]. It is also possible that hydrogen is donated from water and transferred into the biocrude during the degradation process [
49,
50].
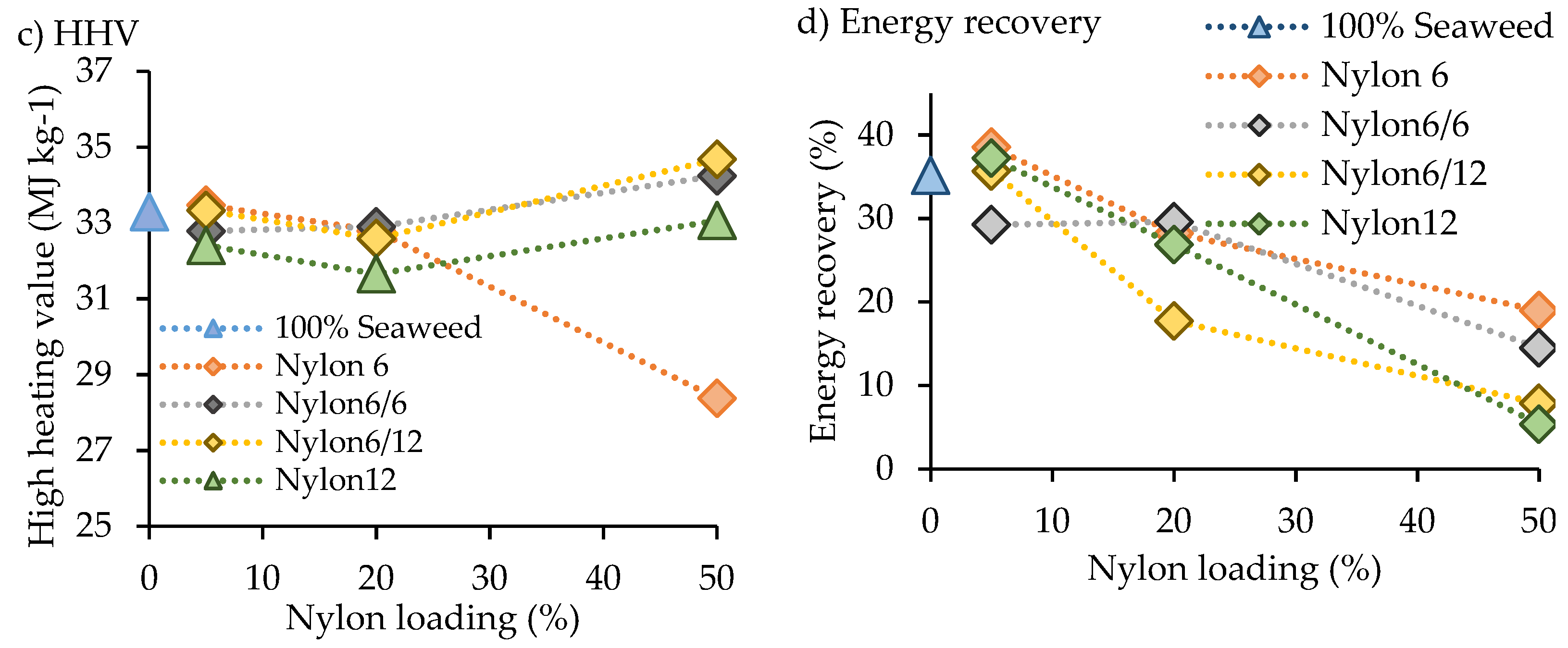
The high heating value (HHV) obtained from pure
F. serratus alone was significantly improved through hydrothermal liquefaction, with a substantial improvement from 9 to 33 MJ kg
−1 for pure macroalgae feedstock and hydrothermal liquefaction of pure
F. serratus, respectively. A similar observation was reported for hydrothermal liquefaction of
Sargassum spp macroalgae biomass [
23]. In the presence of nylon blends, the HHV was not strongly affected, HHV from co-liquefaction of
F. serratus with nylon blends was found to be in the range of 28–35 MJ kg
−1. However, the largest HHV change was found in the presence of 50 wt.% blend nylon 6/12, with a substantial decrease from 33 to 28 MJ kg
−1. This decrease suggests that the biocrude was largely changed by the addition of 50 wt.% nylon 6/12 blend. The highest energy recovery value (38%) was obtained for the biocrude obtained from the addition of 5 wt.% nylon 6 blend, while the lowest energy recovery (5.4%) was obtained from the addition of 50 wt.% nylon 12.
3.4. Solid Residue Composition
Compared to the unprocessed
F. serratus biomass, the quality of the solid residues improved with increased carbon content and high heating value (from 32% to 53% for carbon content and from 9 to 17 MJ kg
−1 for high heating value). The high carbon content of the solid residues resulted from the condensation and carbonization reaction through hydrothermal processing [
51]. The solid residue derived from co-liquefaction with nylon 6 had similar carbon, hydrogen and nitrogen content to solid residue produced from pure macroalgae (
Figure 6). A slight increase in the overall total carbon, nitrogen and hydrogen content of the solid residue produced was observed with the addition of nylon 6/6 blends. Co-liquefaction of nylon 6/12 contributed to an increase in C content (55%, 58% and 63% C for 5, 20 and 50 wt.%, respectively). The addition of nylon 12 in the solid residue gave the highest carbon content and hydrogen content (67% C and 10% H, respectively) suggesting that nylon 12 is the most difficult nylon to breakdown and instead distributes into the solid residue.
For a better understanding of the elemental composition, the classification of solid residue fuel obtained from the HTL process can be illustrated using a Van Krevelen diagram (plot of H/C and O/C atomic ratios). A decrease in H/C and O/C atomic ratio suggests the development in aromatic structure in solid residue due to the removal of hydrogen and oxygen from the original feedstock [
52]. Effective combustion preferably needs lower O/C and H/C ratios as they reduce thermodynamic energy losses, produce less smoke and water vapor [
53,
54].
Figure 7 shows the Van Krevelen diagram for all conditions used in this study and corresponding solid residue. As the HTL progressed, H and O were decreased in the solid residue biochar and became carbon rich. For the co-liquefaction with nylon 6, the overall impact on atomic ratio biochar was similar to that observed from pure macroalgae biochar. This suggests that nylon 6 was found to breakdown completely in the HTL process. In contrast, co-liquefaction with nylon 6/12 and nylon 12 blends contributed to a substantial increase in H/C atomic ratio, while the O/C atomic ratio of the solid residues obtained substantially decreased. This significant difference was increased by a large level of unreacted nylons in the solid residue.
3.5. Yield of Nylon Products from the System
Due to the elevated conversion of nylon 6, and nylon 6/6, the aqueous phase and biocrude produced from the two polymers was also assessed by LC–MS and GC–MS analysis for chemicals produced from the breakdown of the polymers. Since ε-caprolactam was the most produced breakdown compound, found in both the aqueous phase and biocrude, ε-caprolactam was used to estimate the yield of nylon products from the system observed in the aqueous and biocrude products. The quantification of the main compounds present in the aqueous phase and biocrude are shown in
Table 1. The presence of ε-caprolactam was observed in the range of 0.006–0.7 g L
−1 and 0.006.0–1.1 g L
−1 for LC–MS and GC–MS, respectively, which give a high nylon yield conversion of over 80% at 20 wt.% nylon 6 blend. Presence of these compounds suggests that nylon 6 was almost completely decomposed into ε-aminocaproic acid by hydrolysis followed by cyclodehydration to ε-caprolactam [
43]. Co-liquefaction of marine macroalgae with plastic therefore not only produces a suitable product suite for biofuel, but with further separations could yield the nylon monomer for further valorization. Calibration curves and further information are provided in the
Supplementary Material (Figures S5 and S6).
3.6. Conversion of Macroalgae with Nylon Fishing Line
The results have identified the positive correlation between nylon blends and F. serratus for enhancing nylon conversion and biocrude properties. In order to demonstrate the potential benefit in plastic contaminated oceans, ghost fishing line wasted up on shore was co-processed with F. serratus.
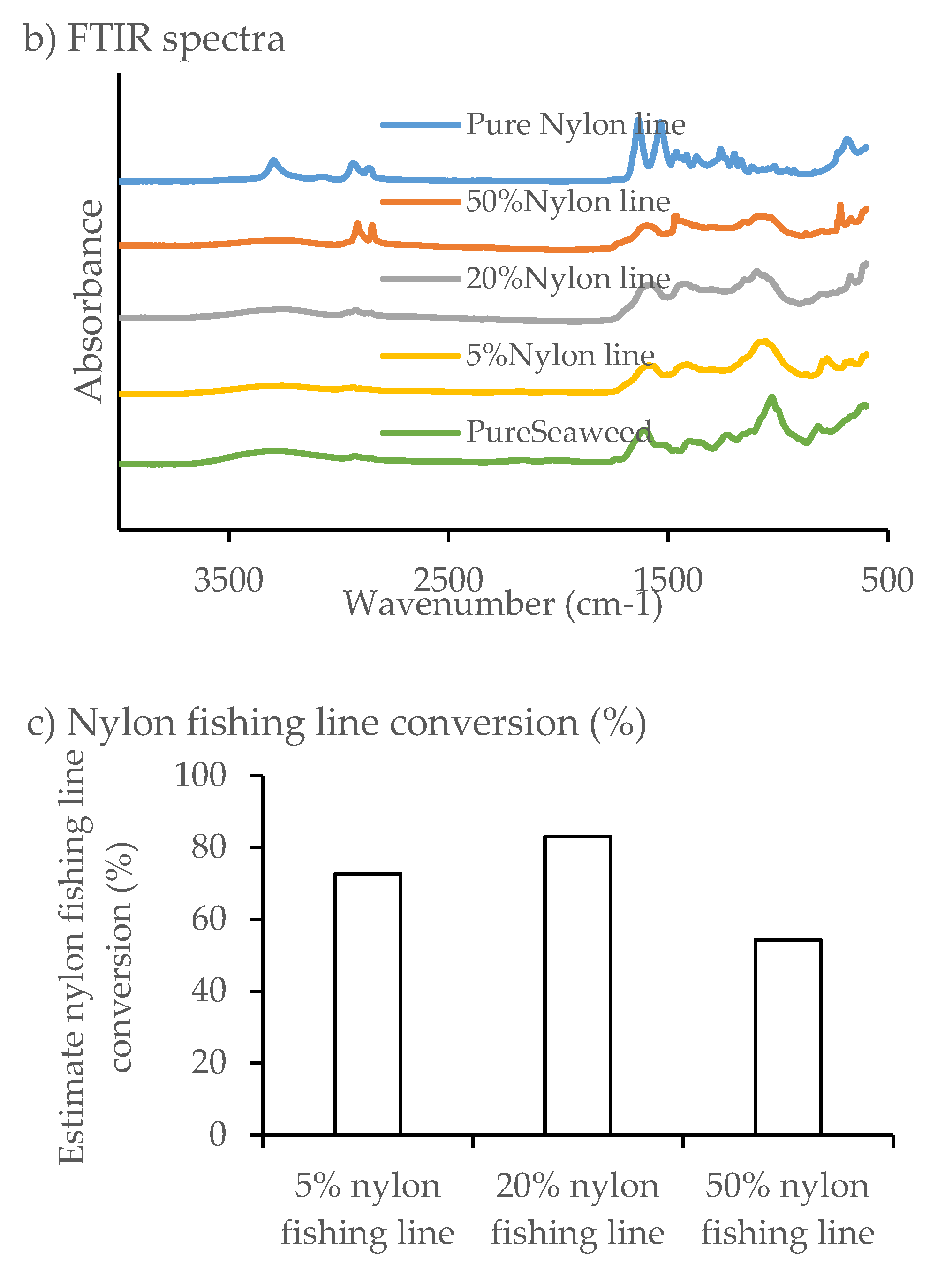
The nylon fishing line was analyzed using FTIR to identify key functional groups. Peaks of intensity were observed at 3300, 2940, 1638 and 1538 cm−1, which were similar to those observed for nylon 6/6.
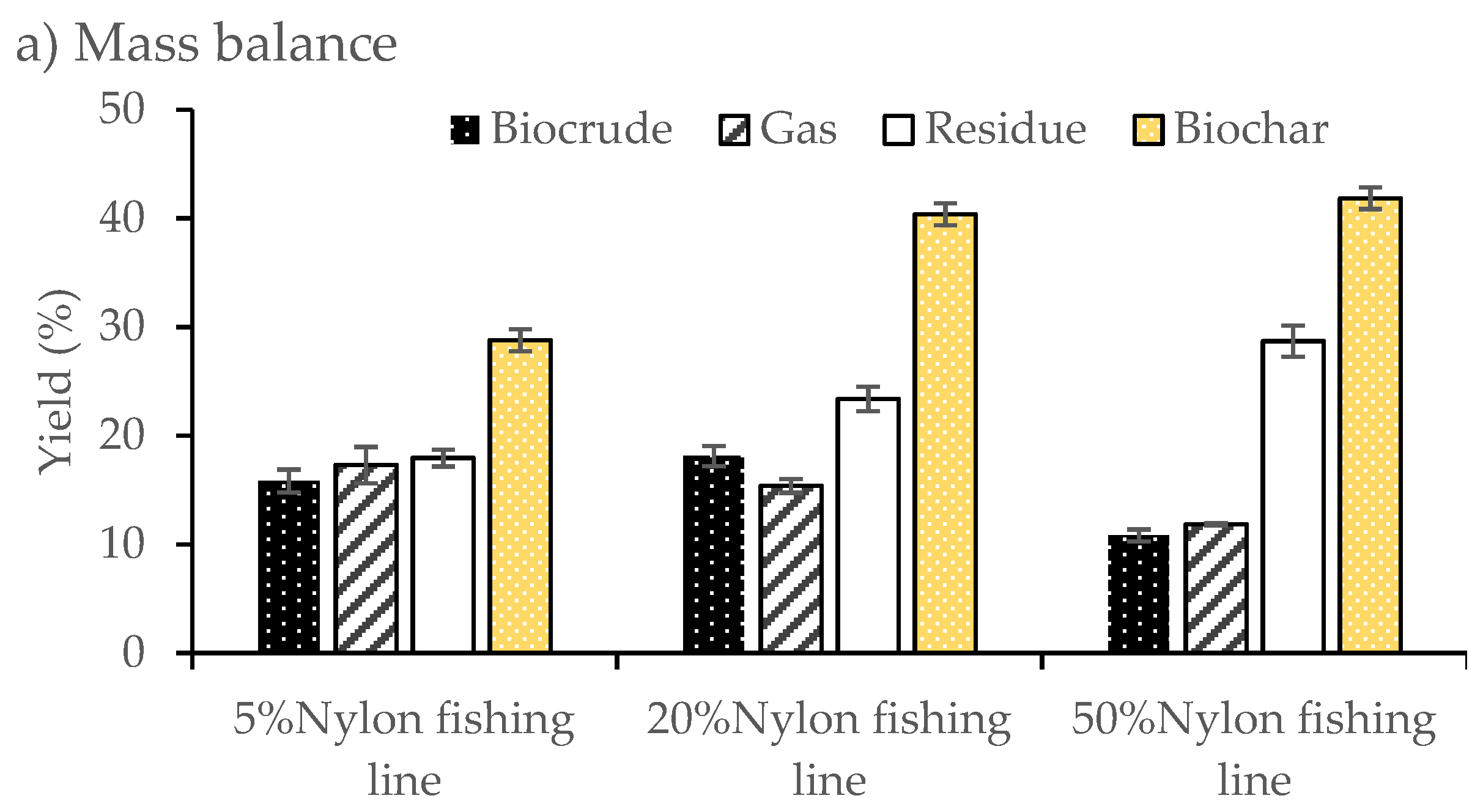
Similar to the model compounds, the HTL of ghost fishing line with
F. serratus was found to increase the biocrude yields compared to pure
F. serratus. A modest increase in biocrude yield was observed for 20 wt.% blend of nylon fishing line blends (18.1%), whilst decreases in overall biocrude production were seen for 5% and 50% blends (biocrude yields of biocrude products of 15.8 and 10.8 wt.%, respectively). Residue aqueous phase product recovery increased steadily (18 wt.% for 5% nylon fishing blend, increased to 28.7 wt.% at a 50% nylon fishing blend), whilst increasing nylon fishing blend levels also caused a modest decrease in the yield of gas product at 50% blend level. The addition of ghost fishing line had effect on the biochar solid residue, biochar increased from 28.8 to 40.4 wt.% and 41.9 wt.% for the 20% and 50% ghost fishing line blend, respectively. These results were similar to those observed for HTL of the nylon 6/6 blend. However, some of the (fishing line) polymer remained unconverted in the HTL reaction, demonstrating a slightly lower overall conversion compared to co-liquefaction of the model nylon 6/6 blends (
Figure 4).
The FTIR spectra of solid residue from HTL of nylon fishing line at 5% and 20% blends and
F. serratus were almost identical to the spectrum of pure macroalgae solid biochar (
Figure 8). The presence of nylon fishing line did not appear to change the solid residue composition, suggesting that nylon fishing line at 5% and 20% blends decomposed almost completely and formed soluble products. In contrast, a number of peaks (intensive peaks at 718, 1475, 2853 and 2923 cm
−1) similar to pure nylon fishing line were observed in the biochar produced from 50% blended nylon fishing line, suggesting the presence of some unreacted nylon fishing line. The interaction between
F. serratus and the nylon fishing line suggests that the presence of
F. serratus reactive fragments affects the thermal stability of nylon fishing line. The presence of metals in
F. serratus ash can enhance the plastic decomposition, which becomes a hydrogen donor [
55], preventing undesirable side reactions and leading to lower solid residue and higher biocrude formation [
56].














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}