Glycyrrhizin Blocks the Detrimental Effects of HMGB1 on Cortical Neurogenesis after Traumatic Neuronal Injury

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
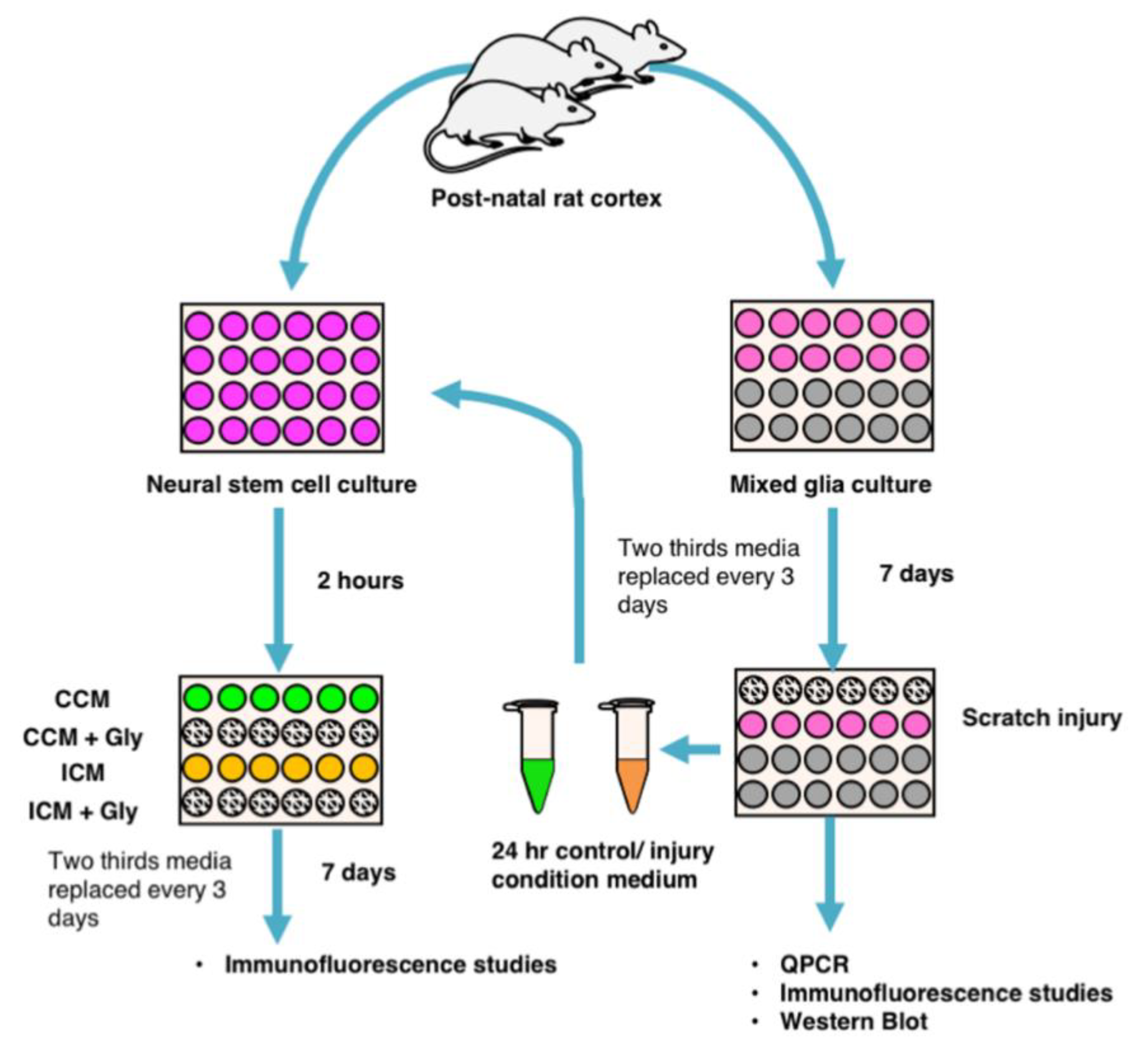
2.1. Generating Mixed Glial Cultures and Injury Condition Media
2.2. Generating NSPC Cultures
2.3. Immunocytochemistry
2.4. Cell Images and Quantification
2.5. Transcriptome Analysis
2.6. PCR Assay
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
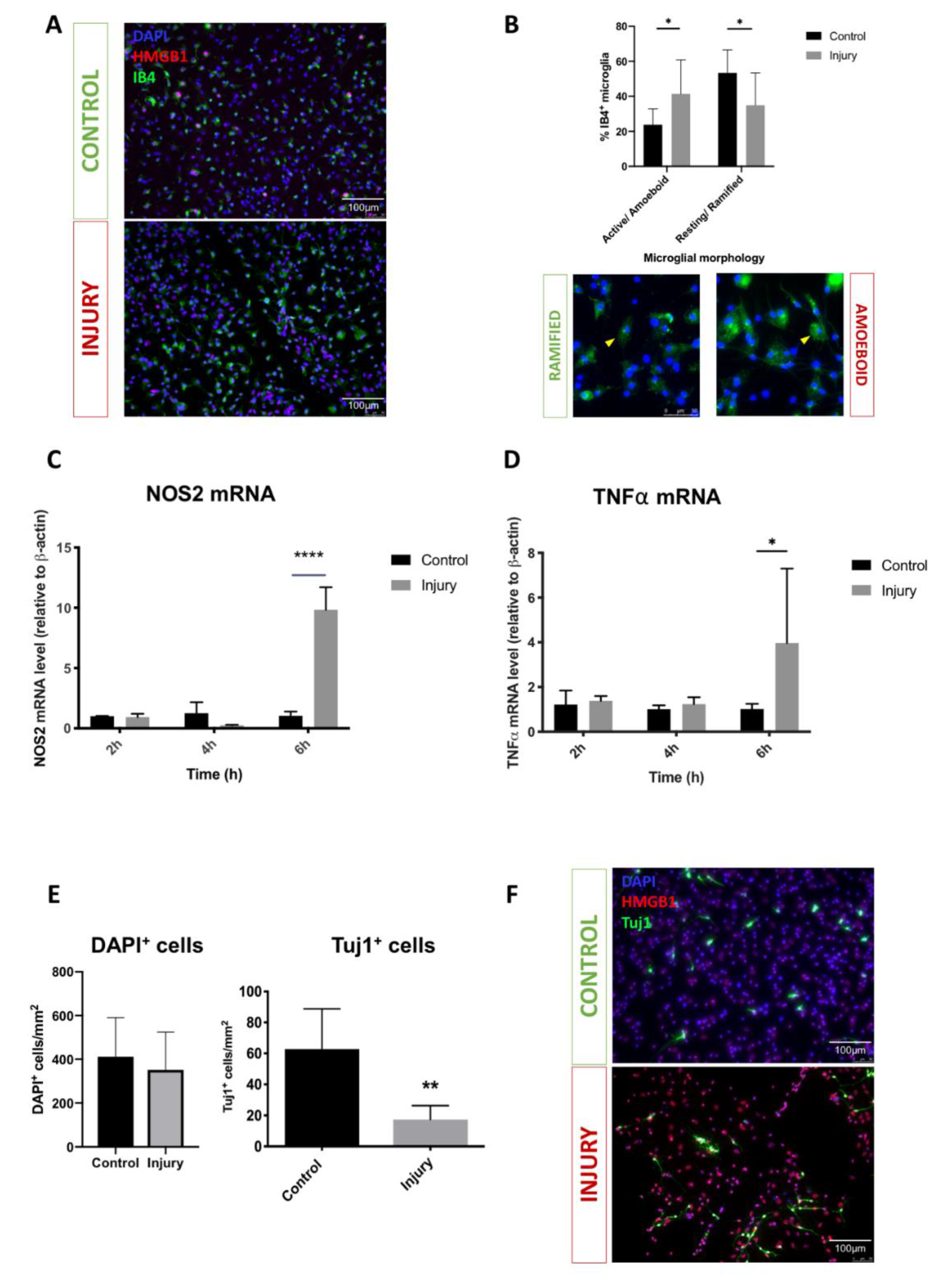
3.1. Mechanical Scratch Injury Enhances Microglial Activation and Neuronal Cell Loss In-Vitro
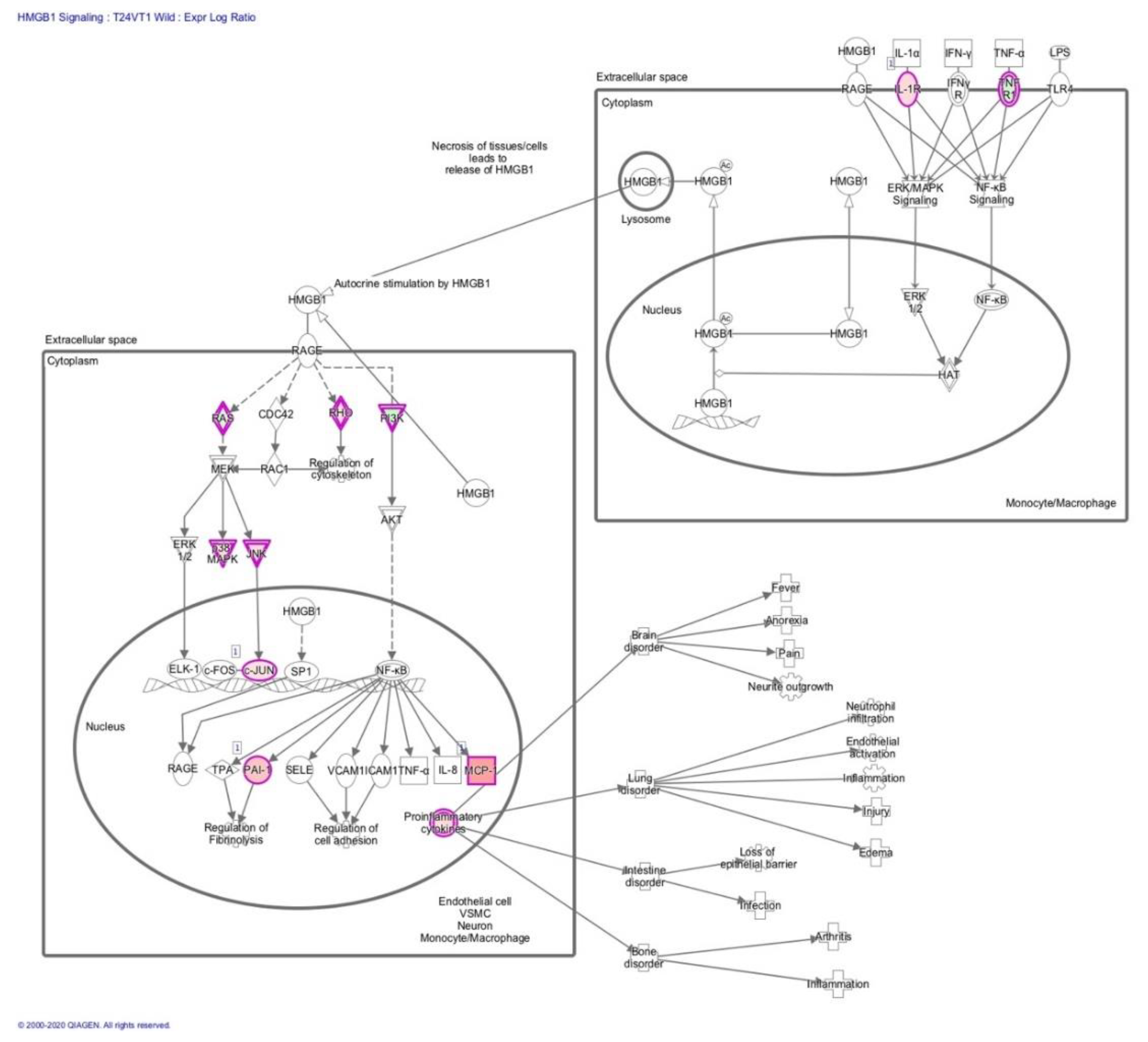
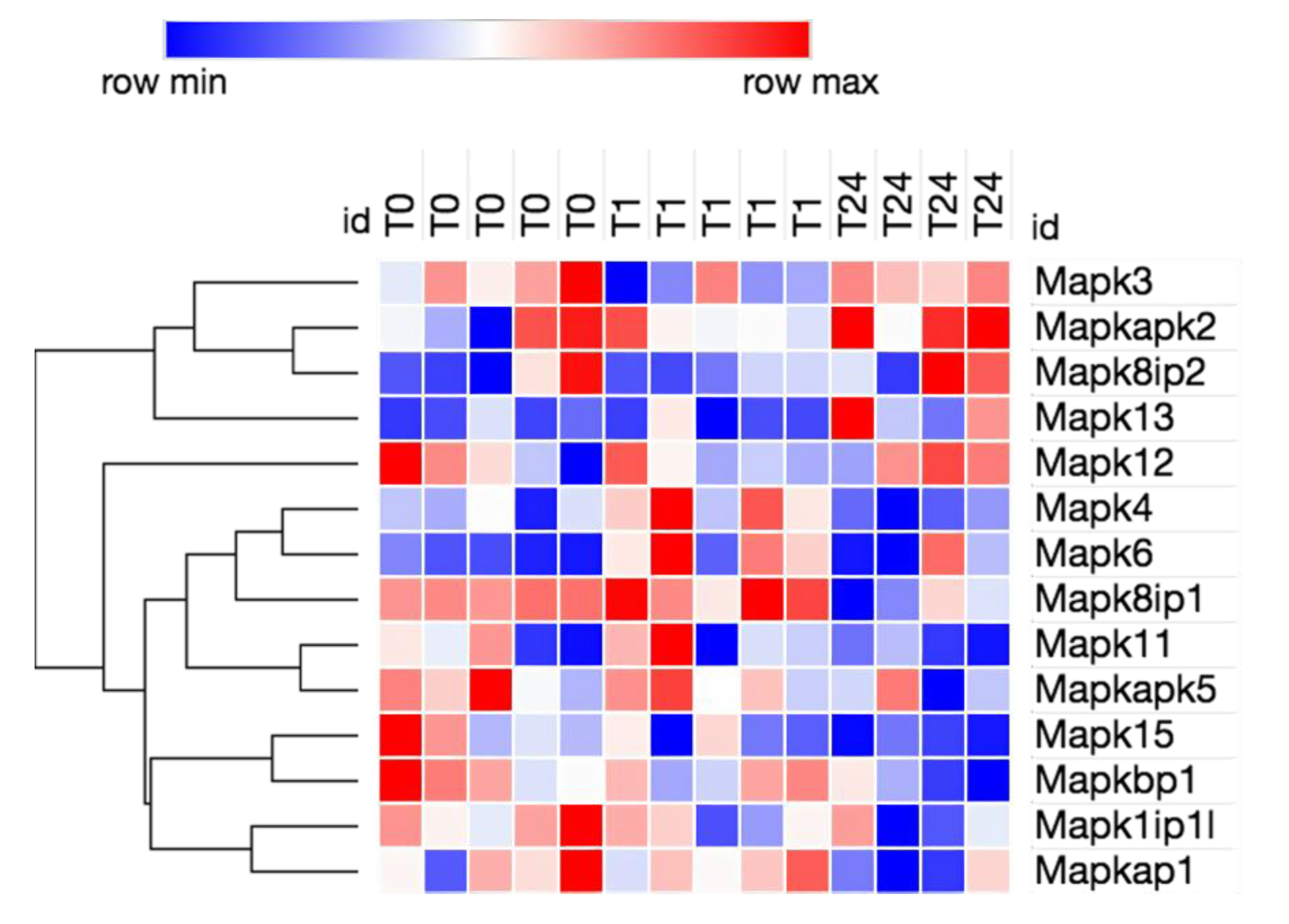
3.2. Transcriptome Analysis Reveals Needle Cortical Injury Upregulate HMGB1 Related Networks In-Vivo
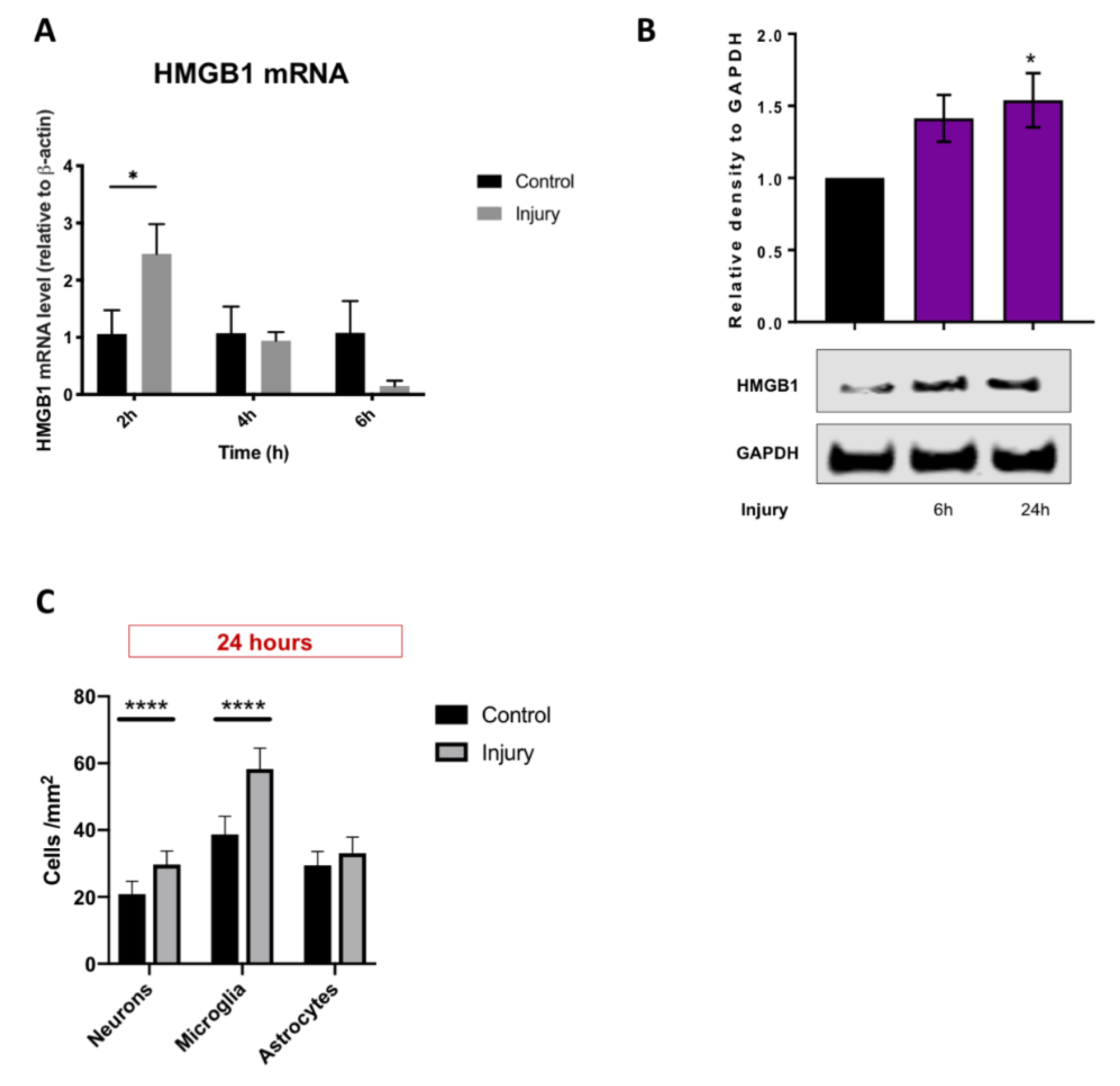
3.3. HMGB1 Cellular Expression and Release Are Increased after Mechanical Scratch Injury In-Vitro
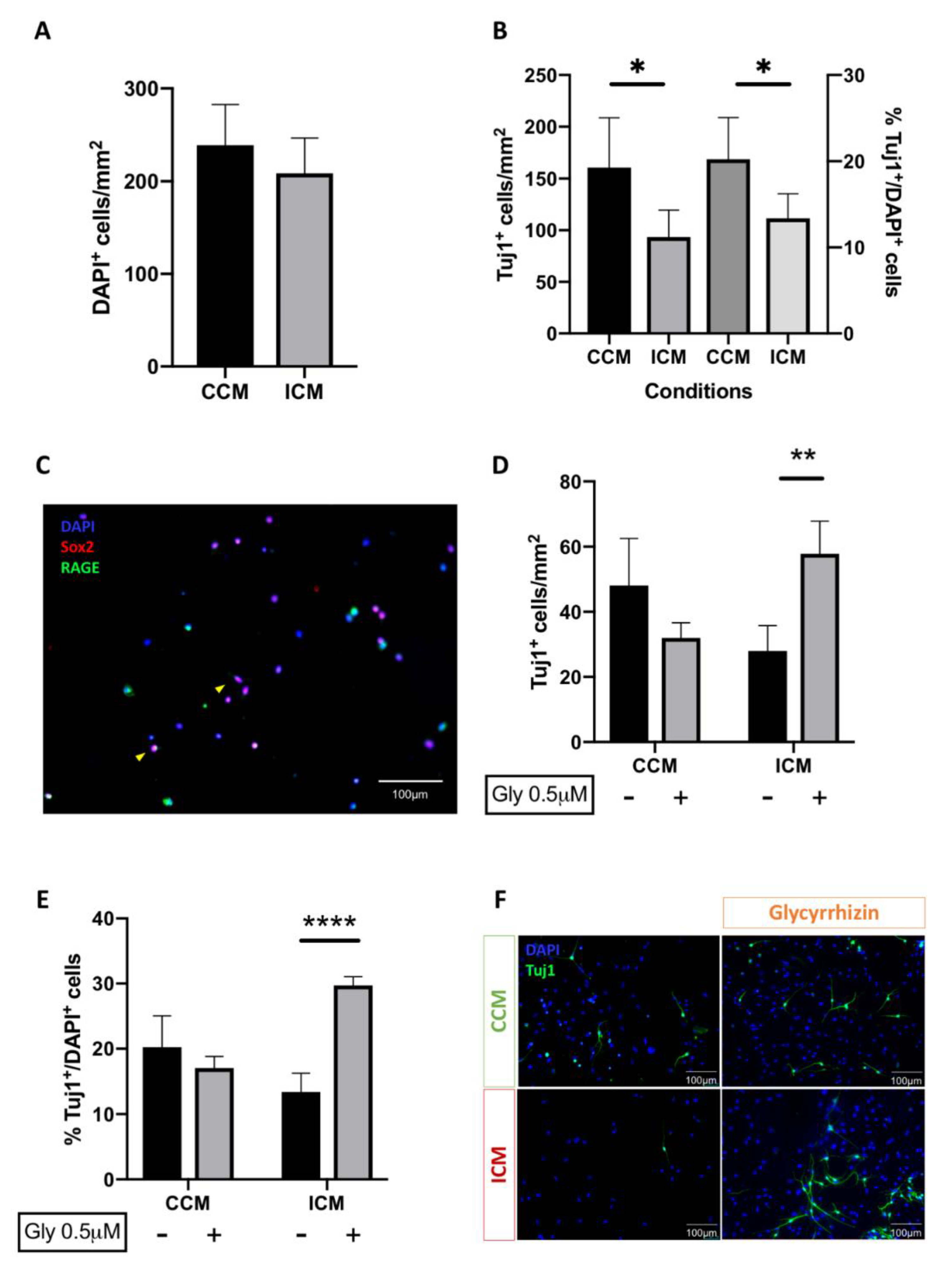
3.4. Injury Conditioned Media Has a Detrimental Effect on Neuronal Progenitor Cell Survival and Differentiation
3.5. Blockade of HMGB1 Receptor Subtype RAGE Mediates ICM Anti-Neurogenic Effects
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Prevalence and most common causes of disability among adults–United States, 2005. Mmwr. Morb. Mortal. Wkly. Rep. 2009, 58, 421–426.
- Andelic, N.; Howe, E.I.; Hellstrom, T.; Sanchez, M.F.; Lu, J.; Lovstad, M.; Roe, C. Disability and quality of life 20 years after traumatic brain injury. Brain Behav. 2018, 8, e01018. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.C.; Hoosain, R.; Lee, T.M.; Fan, Y.W.; Fong, D. Are there sub-types of attentional deficits in patients with persisting post-concussive symptoms? A cluster analytical study. Brain Inj. 2003, 17, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Ricker, J.H.; Hillary, F.G.; DeLuca, J. Functionally activated brain imaging (O-15 PET and fMRI) in the study of learning and memory after traumatic brain injury. J. Head Trauma Rehabil. 2001, 16, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Ghawami, H.; Sadeghi, S.; Raghibi, M.; Rahimi-Movaghar, V. Executive functioning of complicated-mild to moderate traumatic brain injury patients with frontal contusions. Appl. Neuropsychol. Adult 2017, 24, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Drapeau, J.; Gosselin, N.; Peretz, I.; McKerral, M. Emotional recognition from dynamic facial, vocal and musical expressions following traumatic brain injury. Brain Inj. 2017, 31, 221–229. [Google Scholar] [CrossRef]
- Nygren DeBoussard, C.; Lannsjo, M.; Stenberg, M.; Stalnacke, B.M.; Godbolt, A.K. Behavioural problems in the first year after Severe traumatic brain injury: A prospective multicentre study. Clin. Rehabil. 2017, 31, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Lewis, F.D.; Horn, G.J. Depression following traumatic brain injury: Impact on post-hospital residential rehabilitation outcomes. NeuroRehabilitation 2017, 40, 401–410. [Google Scholar] [CrossRef]
- Gao, X.; Chen, J. Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral cortex. J. Neuropathol. Exp. Neurol. 2011, 70, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, H.; Schafer, K.; Hollt, V. BetaIII tubulin-expressing neurons reveal enhanced neurogenesis in hippocampal and cortical structures after a contusion trauma in rats. J. Neurotrauma 2002, 19, 975–983. [Google Scholar] [CrossRef]
- Yi, X.; Jin, G.; Zhang, X.; Mao, W.; Li, H.; Qin, J.; Shi, J.; Dai, K.; Zhang, F. Cortical endogenic neural regeneration of adult rat after traumatic brain injury. PLoS ONE 2013, 8, e70306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsini, A.; Zunszain, P.A.; Thuret, S.; Pariante, C.M. The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci. 2015, 38, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, M.P. Bench-to-bedside review: High-mobility group box 1 and critical illness. Crit. Care (Lond. Engl.) 2007, 11, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 2014, 62, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuma, Y.; Liu, K.; Wake, H.; Liu, R.; Nishimura, Y.; Hui, Z.; Teshigawara, K.; Haruma, J.; Yamamoto, Y.; Yamamoto, H.; et al. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology 2014, 85, 18–26. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Tracey, K.J. Extracellular role of HMGB1 in inflammation and sepsis. J. Intern. Med. 2004, 255, 320–331. [Google Scholar] [CrossRef]
- Au, A.K.; Aneja, R.K.; Bell, M.J.; Bayir, H.; Feldman, K.; Adelson, P.D.; Fink, E.L.; Kochanek, P.M.; Clark, R.S. Cerebrospinal fluid levels of high-mobility group box 1 and cytochrome C predict outcome after pediatric traumatic brain injury. J. Neurotrauma 2012, 29, 2013–2021. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.Y.; Yu, G.F.; Zhang, Z.Y.; Huang, Q.; Dong, X.Q. Plasma high-mobility group box 1 levels and prediction of outcome in patients with traumatic brain injury. Clin. Chim. Acta Int. J. Clin. Chem. 2012, 413, 1737–1741. [Google Scholar] [CrossRef]
- Itoh, T.; Satou, T.; Hashimoto, S.; Ito, H. Isolation of neural stem cells from damaged rat cerebral cortex after traumatic brain injury. Neuroreport 2005, 16, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.I.; Shtaya, A.B.; Zaben, M.J.; Owens, E.V.; Kiecker, C.; Gray, W.P. Endogenous GFAP-positive neural stem/progenitor cells in the postnatal mouse cortex are activated following traumatic brain injury. J. Neurotrauma 2012, 29, 828–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhin, A.G.; Ivanova, S.A.; Knoblach, S.M.; Faden, A.I. New in vitro model of traumatic neuronal injury: Evaluation of secondary injury and glutamate receptor-mediated neurotoxicity. J. Neurotrauma 1997, 14, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Tecoma, E.S.; Monyer, H.; Goldberg, M.P.; Choi, D.W. Traumatic neuronal injury in vitro is attenuated by NMDA antagonists. Neuron 1989, 2, 1541–1545. [Google Scholar] [CrossRef]
- Zaben, M.; Sheward, W.J.; Shtaya, A.; Abbosh, C.; Harmar, A.J.; Pringle, A.K.; Gray, W.P. The neurotransmitter VIP expands the pool of symmetrically dividing postnatal dentate gyrus precursors via VPAC2 receptors or directs them toward a neuronal fate via VPAC1 receptors. Stem Cells 2009, 27, 2539–2551. [Google Scholar] [CrossRef]
- Sun, X.; Zeng, H.; Wang, Q.; Yu, Q.; Wu, J.; Feng, Y.; Deng, P.; Zhang, H. Glycyrrhizin ameliorates inflammatory pain by inhibiting microglial activation-mediated inflammatory response via blockage of the HMGB1-TLR4-NF-kB pathway. Exp. Cell Res. 2018, 369, 112–119. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Lim, C.M.; Yu, Y.M.; Lee, J.K. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J. Neurosci. Res. 2008, 86, 1125–1131. [Google Scholar] [CrossRef]
- Trim Galore. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 11 July 2020).
- FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 11 July 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Ensembl. Available online: http://www.ensembl.org/info/data/ftp/index.html/ (accessed on 11 July 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, A.; Anders, S.; Weatheritt, R.J.; Gibson, T.J.; Steinmetz, L.M.; Huber, W. Drift and conservation of differential exon usage across tissues in primate species. Proc. Natl. Acad. Sci. USA 2013, 110, 15377–15382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiagen. Available online: https://www.qiagen.com/gb/products/discovery-and-translational-research/next-generation-sequencing/informatics-and-data/interpretation-content-databases/ingenuity-pathway-analysis/?clear=true#orderinginformation (accessed on 11 July 2020).
- Morpheus. Available online: https://software.broadinstitute.org/morpheus (accessed on 11 July 2020).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative geneexpression data using real-time quantitative PCR andthe 2[-Delta Delta C(t)] method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Kim, Y.; Cruz, M.O.; Park, E.M.; Chu, C.K.; Song, G.Y.; Joh, T.H. Repression of proinflammatory cytokine and inducible nitric oxide synthase (NOS2) gene expression in activated microglia by N-acetyl-O-methyldopamine: Protein kinase A-dependent mechanism. Glia 2001, 33, 324–333. [Google Scholar] [CrossRef]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Gauley, J.; Pisetsky, D.S. The translocation of HMGB1 during cell activation and cell death. Autoimmunity 2009, 42, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, K.; Mutlak, H.; Smith, W.R.; Shohami, E.; Stahel, P.F. Pharmacology of traumatic brain injury: Where is the “golden bullet”? Mol. Med. 2008, 14, 731–740. [Google Scholar] [CrossRef]
- Leker, R.R.; Shohami, E. Cerebral ischemia and trauma-different etiologies yet similar mechanisms: Neuroprotective opportunities. Brain Res. Brain Res. Rev. 2002, 39, 55–73. [Google Scholar] [CrossRef]
- Ernst, A.; Frisen, J. Adult neurogenesis in humans- common and unique traits in mammals. PLoS Biol. 2015, 13, e1002045. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Jaber, M.; Gaillard, A. Potentials of endogenous neural stem cells in cortical repair. Front Cell Neurosci. 2012, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; ZhuGe, Q.; Zhong, M.; Chen, G.; Shao, B.; Wang, H.; Mao, X.; Xie, L.; Jin, K. Neurogenesis in adult human brain after traumatic brain injury. J. Neurotrauma 2013, 30, 1872–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemirovich-Danchenko, N.M.; Khodanovich, M.Y. New Neurons in the Post-ischemic and Injured Brain: Migrating or Resident? Front. Neurosci. 2019, 13, 588. [Google Scholar] [CrossRef]
- Li, L.; Harms, K.M.; Ventura, P.B.; Lagace, D.C.; Eisch, A.J.; Cunningham, L.A. Focal cerebral ischemia induces a multilineage cytogenic response from adult subventricular zone that is predominantly gliogenic. Glia 2010, 58, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.S.; Overstreet-Wadiche, L. Integration of adult generated neurons during epileptogenesis. Epilepsia 2008, 49 (Suppl. S5), 3–12. [Google Scholar] [CrossRef]
- Faber-Elman, A.; Solomon, A.; Abraham, J.A.; Marikovsky, M.; Schwartz, M. Involvement of wound-associated factors in rat brain astrocyte migratory response to axonal injury: In vitro simulation. J. Clin. Investig. 1996, 97, 162–171. [Google Scholar] [CrossRef]
- Nishio, T.; Kawaguchi, S.; Yamamoto, M.; Iseda, T.; Kawasaki, T.; Hase, T. Tenascin-C regulates proliferation and migration of cultured astrocytes in a scratch wound assay. Neuroscience 2005, 132, 87–102. [Google Scholar] [CrossRef]
- Goldshmit, Y.; Galea, M.P.; Wise, G.; Bartlett, P.F.; Turnley, A.M. Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J. Neurosci. 2004, 24, 10064–10073. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Wu, W.; Hu, Y.C.; Li, H.; Zhang, D.; Li, S.; Li, W.; Li, W.D.; Ma, B.; Zhu, J.H.; et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2014, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Faraco, G.; Fossati, S.; Bianchi, M.E.; Patrone, M.; Pedrazzi, M.; Sparatore, B.; Moroni, F.; Chiarugi, A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J. Neurochem. 2007, 103, 590–603. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rivera, Z.; Jube, S.; Nasu, M.; Bertino, P.; Goparaju, C.; Franzoso, G.; Lotze, M.T.; Krausz, T.; Pass, H.I.; et al. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 12611–12616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef]
- Takizawa, T.; Shibata, M.; Kayama, Y.; Shimizu, T.; Toriumi, H.; Ebine, T.; Unekawa, M.; Koh, A.; Yoshimura, A.; Suzuki, N. High-mobility group box 1 is an important mediator of microglial activation induced by cortical spreading depression. J. Cereb. Blood Flow Metab. 2017, 37, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhang, L.; Teng, J.; Miao, W. HMGB1 mediates microglia activation via the TLR4/NF-kappaB pathway in coriaria lactone induced epilepsy. Mol. Med. Rep. 2018, 17, 5125–5131. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, M.; Monda, A.; Takemoto, R.; Fujimoto, Y.; Sugitani, M.; Iwamura, T.; Hiroyasu, T.; Inoue, A. High-mobility group box 1 up-regulates aquaporin 4 expression via microglia-astrocyte interaction. Neurochem. Int. 2014, 75, 32–38. [Google Scholar] [CrossRef]
- Xue, X.; Chen, X.; Fan, W.; Wang, G.; Zhang, L.; Chen, Z.; Liu, P.; Liu, M.; Zhao, J. High-mobility group box 1 facilitates migration of neural stem cells via receptor for advanced glycation end products signaling pathway. Sci. Rep. 2018, 8, 4513. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Lim, C.M.; Kim, J.B.; Shin, J.H.; Lee, S.; Lee, M.; Lee, J.K. Extracellular HMGB1 released by NMDA treatment confers neuronal apoptosis via RAGE-p38 MAPK/ERK signaling pathway. Neurotox. Res. 2011, 20, 159–169. [Google Scholar] [CrossRef]
- Gu, X.J.; Xu, J.; Ma, B.Y.; Chen, G.; Gu, P.Y.; Wei, D.; Hu, W.X. Effect of glycyrrhizin on traumatic brain injury in rats and its mechanism. Chin. J. Traumatol. Zhonghua Chuang Shang Za Zhi 2014, 17, 1–7. [Google Scholar]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar] [CrossRef] [Green Version]
- Srikrishna, G.; Huttunen, H.J.; Johansson, L.; Weigle, B.; Yamaguchi, Y.; Rauvala, H.; Freeze, H.H. N -Glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J. Neurochem. 2002, 80, 998–1008. [Google Scholar] [CrossRef]
- Simon, D.W.; Aneja, R.K.; Alexander, H.; Bell, M.J.; Bayir, H.; Kochanek, P.M.; Clark, R.S.B. Minocycline Attenuates High Mobility Group Box 1 Translocation, Microglial Activation, and Thalamic Neurodegeneration after Traumatic Brain Injury in Post-Natal Day 17 Rats. J. Neurotrauma 2018, 35, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Lai, W.; Wallace, L.M.; Yang, H.; Popovich, P.G. High mobility group box-1 (HMGB1) is increased in injured mouse spinal cord and can elicit neurotoxic inflammation. Brain Behav. Immun. 2018, 72, 22–33. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Entrez Gene Name | Expr Log Ratio | Expr False Discovery Rate (q-Value) | Expression |
|---|---|---|---|---|
| CCL2 | C-C motif chemokine ligand 2 |  2.991 2.991 | 0.000265 | Up |
| CLCF1 | cardiotrophin like cytokine factor 1 | 1.574 | 0.0353 | |
| IL1R1 | interleukin 1 receptor type 1 | 1.154 | 0.0275 | Down |
| JUN | Jun proto-oncogene, AP-1 transcription factor subunit | 0.814 | 0.00194 | Up |
| MAPK12 | mitogen-activated protein kinase 12 | 1.224 | 0.00468 | Up |
| NGFR | nerve growth factor receptor |  −2.276 −2.276 | 0.0138 | Up |
| PIK3C2G | phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 gamma | −2.42 | 0.0202 | Up |
| RAC2 | Rac family small GTPase 2 | 1.652 | 3.11E-05 | |
| RAC3 | Rac family small GTPase 3 | 0.486 | 0.0391 | |
| RALB | RAS like proto-oncogene B | 0.332 | 0.0212 | Up |
| RASD2 | RASD family member 2 | −1.83 | 1.85E-05 | |
| RHOC | ras homolog family member C | 0.591 | 0.0137 | Up |
| RHOH | ras homolog family member H | 0.872 | 0.0282 | Up |
| RRAS | RAS related | 0.587 | 0.00878 | Up |
| SERPINE1 | serpin family E member 1 | 1.876 | 0.0198 | |
| TGFB1 | transforming growth factor beta 1 | 0.685 | 0.0194 | Up |
| TNFRSF11B | TNF receptor superfamily member 11b | 0.873 | 0.0488 | Up |
| TNFRSF1A | TNF receptor superfamily member 1A | 0.965 | 0.00707 | Up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manivannan, S.; Harari, B.; Muzaffar, M.; Elalfy, O.; Hettipathirannahelage, S.; James, Z.; Sharouf, F.; Ormonde, C.; Alsaqati, M.; Gray, W.; et al. Glycyrrhizin Blocks the Detrimental Effects of HMGB1 on Cortical Neurogenesis after Traumatic Neuronal Injury. Brain Sci. 2020, 10, 760. https://doi.org/10.3390/brainsci10100760
Manivannan S, Harari B, Muzaffar M, Elalfy O, Hettipathirannahelage S, James Z, Sharouf F, Ormonde C, Alsaqati M, Gray W, et al. Glycyrrhizin Blocks the Detrimental Effects of HMGB1 on Cortical Neurogenesis after Traumatic Neuronal Injury. Brain Sciences. 2020; 10(10):760. https://doi.org/10.3390/brainsci10100760
Chicago/Turabian StyleManivannan, Susruta, Balkis Harari, Maryam Muzaffar, Omar Elalfy, Sameera Hettipathirannahelage, Zoe James, Feras Sharouf, Chloe Ormonde, Mouhamed Alsaqati, William Gray, and et al. 2020. "Glycyrrhizin Blocks the Detrimental Effects of HMGB1 on Cortical Neurogenesis after Traumatic Neuronal Injury" Brain Sciences 10, no. 10: 760. https://doi.org/10.3390/brainsci10100760
APA StyleManivannan, S., Harari, B., Muzaffar, M., Elalfy, O., Hettipathirannahelage, S., James, Z., Sharouf, F., Ormonde, C., Alsaqati, M., Gray, W., & Zaben, M. (2020). Glycyrrhizin Blocks the Detrimental Effects of HMGB1 on Cortical Neurogenesis after Traumatic Neuronal Injury. Brain Sciences, 10(10), 760. https://doi.org/10.3390/brainsci10100760