Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease


Abstract
:
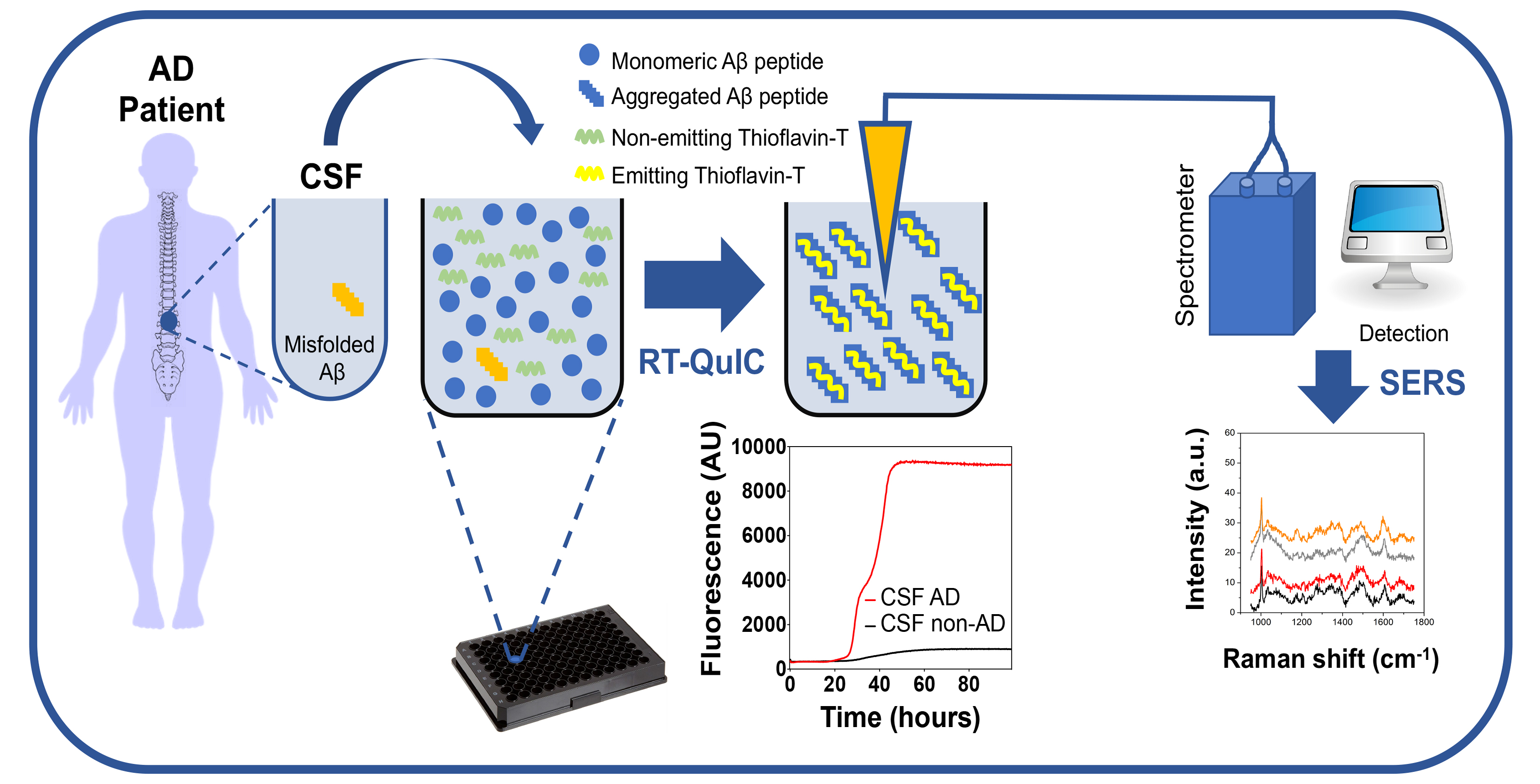{kind=link}
1. Alzheimer’s Disease is a Double-Prion Disorder
2. Phenotypic Heterogeneity of Alzheimer’s Disease and the Role of Aβ Strains
3. Challenges in the Clinical Diagnosis of Alzheimer’s Disease
4. Alzheimer’s Disease Biomarkers: The Role of CSF and Other Peripheral Tissues
5. Technological Innovations and the Future of Alzheimer’s Disease Biomarker Discovery
6. Our Contribution to the Field of Alzheimer’s Disease Diagnosis
Author Contributions
Funding
Conflicts of Interest
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef]
- 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [CrossRef]
- Swearer, J.M.; O’Donnell, B.F.; Ingram, S.M.; Drachman, D.A. Rate of progression in familial Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 1996, 9, 22–25. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Review Article: Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers. Res. Ther. 2010, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLOS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.-Y.R.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-Onset Familial Alzheimer’s Disease (EOFAD). Can. J. Neurol. Sci. J. Can. des Sci. Neurol. 2012, 39, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.S.; Tangalos, E.G.; Petersen, R.C.; Smith, G.E.; Schaid, D.J.; Kokmen, E.; Ivnik, R.J.; Thibodeau, S.N. Apolipoprotein E: Risk factor for Alzheimer disease. Am. J. Hum. Genet. 1994, 54, 643–649. [Google Scholar]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Kelényi, G. Thioflavin S fluorescent and congo red anisotropic stainings in the histologic demonstration of amyloid. Acta Neuropathol. 1967, 7, 336–348. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 608–655. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C. Prion-like mechanisms in Alzheimer disease. In Handbook of Clinical Neurology; Elsevier BV: Amsterdam, The Netherlands, 2018; Volume 153, pp. 303–319. [Google Scholar]
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010, 33, 317–325. [Google Scholar] [CrossRef]
- Morales, R.; Callegari, K.; Soto, C. Prion-like features of misfolded Aβ and tau aggregates. Virus Res. 2015, 207, 106–112. [Google Scholar] [CrossRef]
- Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [Green Version]
- CRICK, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Bendheim, P.E.; Brown, H.R.; Rudelli, R.D.; Scala, L.J.; Goller, N.L.; Wen, G.Y.; Kascsak, R.J.; Cashman, N.R.; Bolton, D.C. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 1992, 42, 149. [Google Scholar] [CrossRef]
- Dee, D.R.; Woodside, M.T. Comparing the energy landscapes for native folding and aggregation of PrP. Prion 2016, 10, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Gambetti, P.; Kong, Q.; Zou, W.; Parchi, P.; Chen, S.G. Sporadic and familial CJD: Classification and characterisation. Br. Med. Bull. 2003, 66, 213–239. [Google Scholar] [CrossRef] [Green Version]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.C.; Jucker, M. Neurodegenerative Diseases: Expanding the Prion Concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Ma, J.; Kordower, J.H. How strong is the evidence that Parkinsonʼs disease is a prion disorder? Curr. Opin. Neurol. 2016, 29, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Vasili, E.; Dominguez-Meijide, A.; Outeiro, T.F. Spreading of α-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front. Mol. Neurosci. 2019, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Pecho-Vrieseling, E.; Rieker, C.; Fuchs, S.; Bleckmann, D.; Esposito, M.S.; Botta, P.; Goldstein, C.; Bernhard, M.; Galimberti, I.; Müller, M.; et al. Transneuronal propagation of mutant huntingtin contributes to non–cell autonomous pathology in neurons. Nat. Neurosci. 2014, 17, 1064–1072. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. A Unifying Role for Prions in Neurodegenerative Diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl. Cell Differ. 2011, 53, 565–576. [Google Scholar]
- Raina, A.K.; Monteiro, M.J.; McShea, A.; Smith, M.A. The role of cell cycle-mediated events in Alzheimer’s disease. Int. J. Exp. Pathol. 1999, 80, 71–76. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, L.; Tsvetanov, K.A.; Jones, P.S.; Bevan-Jones, W.R.; Arnold, R.; Borchert, R.J.; Mak, E.; Su, L.; O’Brien, J.T.; Rowe, J.B. Neuroinflammation and Functional Connectivity in Alzheimer’s Disease: Interactive Influences on Cognitive Performance. J. Neurosci. 2019, 39, 7218–7226. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers. Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Lau, H.H.C.; Ingelsson, M.; Watts, J.C. The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol. 2020, 1–23. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Suttkus, A.; Holzer, M.; Morawski, M.; Arendt, T. The neuronal extracellular matrix restricts distribution and internalization of aggregated Tau-protein. Neuroscience 2016, 313, 225–235. [Google Scholar] [CrossRef]
- Warren, J.D.; Fletcher, P.D.; Golden, H.L. The paradox of syndromic diversity in Alzheimer disease. Nat. Rev. Neurol. 2012, 8, 451–464. [Google Scholar] [CrossRef]
- Suárez-González, A.; Henley, S.M.; Walton, J.; Crutch, S.J. Posterior Cortical Atrophy. Psychiatr. Clin. North Am. 2015, 38, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villain, N.; Dubois, B. Alzheimer’s Disease Including Focal Presentations. Semin. Neurol. 2019, 39, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Maderna, E.; Ghidoni, R.; Benussi, L.; Tonoli, E.; Giaccone, G.; Moda, F.; Paterlini, A.; Campagnani, I.; et al. Molecular subtypes of Alzheimer’s disease. Sci. Rep. 2018, 8, 3269. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D. Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3081. [Google Scholar] [CrossRef] [Green Version]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Zhang, J.; Liu, Q.; Liu, C.-C.; Zhang, L.; Bu, G. Heparan Sulphate Proteoglycan and the Low-Density Lipoprotein Receptor-Related Protein 1 Constitute Major Pathways for Neuronal Amyloid- Uptake. J. Neurosci. 2011, 31, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, J.; Mahler, J.; Beschorner, N.; Kaeser, S.A.; Häsler, L.M.; Baumann, F.; Nyström, S.; Portelius, E.; Blennow, K.; Lashley, T.; et al. Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 13018–13023. [Google Scholar] [CrossRef] [Green Version]
- Condello, C.; Lemmin, T.; Stöhr, J.; Nick, M.; Wu, Y.; Maxwell, A.M.; Watts, J.C.; Caro, C.D.; Oehler, A.; Keene, C.D.; et al. Structural heterogeneity and intersubject variability of Aβ in familial and sporadic Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E782–E791. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Appleby, B.; Safar, J.G. Distinct prion-like strains of amyloid beta implicated in phenotypic diversity of Alzheimer’s disease. Prion 2016, 10, 9–17. [Google Scholar] [CrossRef] [Green Version]
- LeVine, H.; Walker, L.C. Molecular polymorphism of Aβ in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Piccini, A.; Russo, C.; Gliozzi, A.; Relini, A.; Vitali, A.; Borghi, R.; Giliberto, L.; Armirotti, A.; D’Arrigo, C.; Bachi, A.; et al. β-Amyloid Is Different in Normal Aging and in Alzheimer Disease. J. Biol. Chem. 2005, 280, 34186–34192. [Google Scholar] [CrossRef] [Green Version]
- Maarouf, C.L.; Daugs, I.D.; Spina, S.; Vidal, R.; Kokjohn, T.A.; Patton, R.L.; Kalback, W.M.; Luehrs, D.C.; Walker, D.G.; Castaño, E.M.; et al. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol. Neurodegener. 2008, 3, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, K.P.R.; Åslund, A.; Berg, I.; Nyström, S.; Konradsson, P.; Herland, A.; Inganäs, O.; Stabo-Eeg, F.; Lindgren, M.; Westermark, G.T.; et al. Imaging Distinct Conformational States of Amyloid-β Fibrils in Alzheimer’s Disease Using Novel Luminescent Probes. ACS Chem. Biol. 2007, 2, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Ghetti, B. Characterization of Amyloid Deposits in Neurodegenerative Diseases. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; pp. 241–258. ISBN 9781617793271. [Google Scholar]
- Steinerman, J.R.; Irizarry, M.; Scarmeas, N.; Raju, S.; Brandt, J.; Albert, M.; Blacker, D.; Hyman, B.; Stern, Y. Distinct Pools of β-Amyloid in Alzheimer Disease–Affected Brain. Arch. Neurol. 2008, 65, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Condello, C.; Stohr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of A prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef] [Green Version]
- Stohr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (A) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef] [Green Version]
- Heilbronner, G.; Eisele, Y.S.; Langer, F.; Kaeser, S.A.; Novotny, R.; Nagarathinam, A.; Åslund, A.; Hammarström, P.; Nilsson, K.P.R.; Jucker, M. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013, 14, 1017–1022. [Google Scholar] [CrossRef] [Green Version]
- Stohr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic A prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Riquelme, A.; Lau, H.H.C.; Stuart, E.; Goczi, A.N.; Wang, Z.; Schmitt-Ulms, G.; Watts, J.C. Prion-like propagation of β-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol. Commun. 2018, 6, 26. [Google Scholar] [CrossRef]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for Seeding of β-Amyloid by Intracerebral Infusion of Alzheimer Brain Extracts in β-Amyloid Precursor Protein-Transgenic Mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Luehmann, M. Exogenous Induction of Cerebral -Amyloidogenesis Is Governed by Agent and Host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Masters, C.L.; Selkoe, D.J. Biochemistry of Amyloid -Protein and Amyloid Deposits in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- Atarashi, R.; Sano, K.; Satoh, K.; Nishida, N. Real-time quaking-induced conversion. Prion 2011, 5, 150–153. [Google Scholar] [CrossRef] [Green Version]
- Paravastu, A.K.; Qahwash, I.; Leapman, R.D.; Meredith, S.C.; Tycko, R. Seeded growth of -amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc. Natl. Acad. Sci. USA 2009, 106, 7443–7448. [Google Scholar] [CrossRef] [Green Version]
- Petersen, R.C.; Caracciolo, B.; Brayne, C.; Gauthier, S.; Jelic, V.; Fratiglioni, L. Mild cognitive impairment: A concept in evolution. J. Intern. Med. 2014, 275, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Ferman, T.J.; Smith, G.E.; Kantarci, K.; Boeve, B.F.; Pankratz, V.S.; Dickson, D.W.; Graff-Radford, N.R.; Wszolek, Z.; Van Gerpen, J.; Uitti, R.; et al. Nonamnestic mild cognitive impairment progresses to dementia with Lewy bodies. Neurology 2013, 81, 2032–2038. [Google Scholar] [CrossRef] [Green Version]
- de Mendonça, A.; Ribeiro, F.; Guerreiro, M.; Garcia, C. Frontotemporal mild cognitive impairment. J. Alzheimers. Dis. 2004, 6, 1–9. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, M.N.; Lue, L.-F.; Fayard, D.; Shi, J. Increasing Precision of Clinical Diagnosis of Alzheimer’s Disease Using a Combined Algorithm Incorporating Clinical and Novel Biomarker Data. Neurol. Ther. 2017, 6, 83–95. [Google Scholar] [CrossRef]
- Niemantsverdriet, E.; Valckx, S.; Bjerke, M.; Engelborghs, S. Alzheimer’s disease CSF biomarkers: Clinical indications and rational use. Acta Neurol. Belg. 2017, 117, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Engelborghs, S.; Niemantsverdriet, E.; Struyfs, H.; Blennow, K.; Brouns, R.; Comabella, M.; Dujmovic, I.; Flier, W.; Frölich, L.; Galimberti, D.; et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit. 2017, 8, 111–126. [Google Scholar] [CrossRef]
- Catania, M.; Di Fede, G.; Tonoli, E.; Benussi, L.; Pasquali, C.; Giaccone, G.; Maderna, E.; Ghidoni, R.; Tagliavini, F. Mirror Image of the Amyloid-β Species in Cerebrospinal Fluid and Cerebral Amyloid in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 877–881. [Google Scholar] [CrossRef]
- Strozyk, D.; Blennow, K.; White, L.R.; Launer, L.J. CSF Aβ 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 2003, 60, 652–656. [Google Scholar] [CrossRef]
- Fagan, A.M.; Mintun, M.A.; Mach, R.H.; Lee, S.-Y.; Dence, C.S.; Shah, A.R.; LaRossa, G.N.; Spinner, M.L.; Klunk, W.E.; Mathis, C.A.; et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ 42 in humans. Ann. Neurol. 2006, 59, 512–519. [Google Scholar] [CrossRef]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.-K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal Fluid β-Amyloid 42 and Tau Proteins as Biomarkers of Alzheimer-Type Pathologic Changes in the Brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Portelius, E.; Tran, A.J.; Andreasson, U.; Persson, R.; Brinkmalm, G.; Zetterberg, H.; Blennow, K.; Westman-Brinkmalm, A. Characterization of Amyloid β Peptides in Cerebrospinal Fluid by an Automated Immunoprecipitation Procedure Followed by Mass Spectrometry. J. Proteome Res. 2007, 6, 4433–4439. [Google Scholar] [CrossRef]
- Biscetti, L.; Salvadori, N.; Farotti, L.; Cataldi, S.; Eusebi, P.; Paciotti, S.; Parnetti, L. The added value of Aβ42/Aβ40 in the CSF signature for routine diagnostics of Alzheimer’s disease. Clin. Chim. Acta 2019, 494, 71–73. [Google Scholar] [CrossRef]
- Shahpasand-Kroner, H.; Klafki, H.-W.; Bauer, C.; Schuchhardt, J.; Hüttenrauch, M.; Stazi, M.; Bouter, C.; Wirths, O.; Vogelgsang, J.; Wiltfang, J. A two-step immunoassay for the simultaneous assessment of Aβ38, Aβ40 and Aβ42 in human blood plasma supports the Aβ42/Aβ40 ratio as a promising biomarker candidate of Alzheimer’s disease. Alzheimers. Res. Ther. 2018, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Lewczuk, P.; Lelental, N.; Spitzer, P.; Maler, J.M.; Kornhuber, J. Amyloid-β 42/40 Cerebrospinal Fluid Concentration Ratio in the Diagnostics of Alzheimer’s Disease: Validation of Two Novel Assays. J. Alzheimer’s Dis. 2014, 43, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Matzen, A.; Blennow, K.; Parnetti, L.; Molinuevo, J.L.; Eusebi, P.; Kornhuber, J.; Morris, J.C.; Fagan, A.M. Cerebrospinal Fluid Aβ42/40 Corresponds Better than Aβ42 to Amyloid PET in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 55, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janelidze, S.; Zetterberg, H.; Mattsson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF A β 42/A β 40 and A β 42/A β 38 ratios: Better diagnostic markers of Alzheimer disease. Ann. Clin. Transl. Neurol. 2016, 3, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Savva, G.M.; Wharton, S.B.; Ince, P.G.; Forster, G.; Matthews, F.E.; Brayne, C. Age, Neuropathology, and Dementia. N. Engl. J. Med. 2009, 360, 2302–2309. [Google Scholar] [CrossRef]
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimer’s Dement. 2018, 14, 1470–1481. [Google Scholar] [CrossRef]
- Wallin, A.K.; Blennow, K.; Zetterberg, H.; Londos, E.; Minthon, L.; Hansson, O. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology 2010, 74, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Zetterberg, H.; Mattsson, N.; Johansson, P.; Minthon, L.; Blennow, K.; Olsson, M.; Hansson, O. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 2015, 85, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Wallin, A.; Ågren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. tau protein in cerebrospinal fluid. Mol. Chem. Neuropathol. 1995, 26, 231–245. [Google Scholar] [CrossRef]
- Buerger, K.; Zinkowski, R.; Teipel, S.J.; Tapiola, T.; Arai, H.; Blennow, K.; Andreasen, N.; Hofmann-Kiefer, K.; DeBernardis, J.; Kerkman, D.; et al. Differential Diagnosis of Alzheimer Disease With Cerebrospinal Fluid Levels of Tau Protein Phosphorylated at Threonine 231. Arch. Neurol. 2002, 59, 1267. [Google Scholar] [CrossRef]
- Riemenschneider, M.; Wagenpfeil, S.; Vanderstichele, H.; Otto, M.; Wiltfang, J.; Kretzschmar, H.; Vanmechelen, E.; Förstl, H.; Kurz, A. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt–Jakob disease from other dementias. Mol. Psychiatry 2003, 8, 343–347. [Google Scholar] [CrossRef]
- Bjerke, M.; Andreasson, U.; Kuhlmann, J.; Portelius, E.; Pannee, J.; Lewczuk, P.; Umek, R.M.; Vanmechelen, E.; Vanderstichele, H.; Stoops, E.; et al. Assessing the commutability of reference material formats for the harmonization of amyloid-β measurements. Clin. Chem. Lab. Med. 2016, 54, 1177–1191. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, N.; Andreasson, U.; Persson, S.; Carrillo, M.C.; Collins, S.; Chalbot, S.; Cutler, N.; Dufour-Rainfray, D.; Fagan, A.M.; Heegaard, N.H.H.; et al. CSF biomarker variability in the Alzheimer’s Association quality control program. Alzheimers. Dement. 2013, 9, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef]
- Engelborghs, S.; De Vreese, K.; Van de Casteele, T.; Vanderstichele, H.; Van Everbroeck, B.; Cras, P.; Martin, J.-J.; Vanmechelen, E.; De Deyn, P.P. Diagnostic performance of a CSF-biomarker panel in autopsy-confirmed dementia. Neurobiol. Aging 2008, 29, 1143–1159. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Jenkins, R.; Watt, H.C.; Green, A.J.E.; Thompson, E.J.; Keir, G.; Fox, N.C.; Rossor, M.N. Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer’s disease. Neurosci. Lett. 2003, 336, 167–170. [Google Scholar] [CrossRef]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020, 11, 1683. [Google Scholar] [CrossRef] [Green Version]
- Cicognola, C.; Brinkmalm, G.; Wahlgren, J.; Portelius, E.; Gobom, J.; Cullen, N.C.; Hansson, O.; Parnetti, L.; Constantinescu, R.; Wildsmith, K.; et al. Novel tau fragments in cerebrospinal fluid: Relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, M.; Blomberg, M.; Jonsson, M.; Wahlund, L.O.; Edman, A.; Lind, K.; Rosengren, L.; Blennow, K.; Wallin, A. Neurofilament protein in cerebrospinal fluid: A marker of white matter changes. J. Neurosci. Res. 2001, 66, 510–516. [Google Scholar] [CrossRef]
- Idland, A.-V.; Sala-Llonch, R.; Borza, T.; Watne, L.O.; Wyller, T.B.; Brækhus, A.; Zetterberg, H.; Blennow, K.; Walhovd, K.B.; Fjell, A.M. CSF neurofilament light levels predict hippocampal atrophy in cognitively healthy older adults. Neurobiol. Aging 2017, 49, 138–144. [Google Scholar] [CrossRef]
- Zetterberg, H.; Skillbäck, T.; Mattsson, N.; Trojanowski, J.Q.; Portelius, E.; Shaw, L.M.; Weiner, M.W.; Blennow, K. Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol. 2016, 73, 60. [Google Scholar] [CrossRef]
- Thorsell, A.; Bjerke, M.; Gobom, J.; Brunhage, E.; Vanmechelen, E.; Andreasen, N.; Hansson, O.; Minthon, L.; Zetterberg, H.; Blennow, K. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010, 1362, 13–22. [Google Scholar] [CrossRef]
- Tarawneh, R.; D’Angelo, G.; Crimmins, D.; Herries, E.; Griest, T.; Fagan, A.M.; Zipfel, G.J.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. Diagnostic and Prognostic Utility of the Synaptic Marker Neurogranin in Alzheimer Disease. JAMA Neurol. 2016, 73, 561. [Google Scholar] [CrossRef]
- De Vos, A.; Jacobs, D.; Struyfs, H.; Fransen, E.; Andersson, K.; Portelius, E.; Andreasson, U.; De Surgeloose, D.; Hernalsteen, D.; Sleegers, K.; et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- De Vos, A.; Struyfs, H.; Jacobs, D.; Fransen, E.; Klewansky, T.; De Roeck, E.; Robberecht, C.; Van Broeckhoven, C.; Duyckaerts, C.; Engelborghs, S.; et al. The Cerebrospinal Fluid Neurogranin/BACE1 Ratio is a Potential Correlate of Cognitive Decline in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 53, 1523–1538. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Blennow, K.; Doody, R.; Hendrix, S.; Lovestone, S.; Salloway, S.; Schindler, R.; Weiner, M.; Zetterberg, H.; Aisen, P.; et al. Plasma Biomarkers of AD Emerging as Essential Tools for Drug Development: An EU/US CTAD Task Force Report. J. Prev. Alzheimer’s Dis. 2019, 6, 169–173. [Google Scholar]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Ovod, V.; Ramsey, K.N.; Mawuenyega, K.G.; Bollinger, J.G.; Hicks, T.; Schneider, T.; Sullivan, M.; Paumier, K.; Holtzman, D.M.; Morris, J.C.; et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer’s Dement. 2017, 13, 841–849. [Google Scholar] [CrossRef]
- Zetterberg, H.; Wilson, D.; Andreasson, U.; Minthon, L.; Blennow, K.; Randall, J.; Hansson, O. Plasma tau levels in Alzheimer’s disease. Alzheimers. Res. Ther. 2013, 5, 9. [Google Scholar] [CrossRef]
- Mielke, M.M.; Hagen, C.E.; Wennberg, A.M.V.; Airey, D.C.; Savica, R.; Knopman, D.S.; Machulda, M.M.; Roberts, R.O.; Jack, C.R.; Petersen, R.C.; et al. Association of Plasma Total Tau Level With Cognitive Decline and Risk of Mild Cognitive Impairment or Dementia in the Mayo Clinic Study on Aging. JAMA Neurol. 2017, 74, 1073. [Google Scholar] [CrossRef]
- Pase, M.P.; Beiser, A.S.; Himali, J.J.; Satizabal, C.L.; Aparicio, H.J.; DeCarli, C.; Chêne, G.; Dufouil, C.; Seshadri, S. Assessment of Plasma Total Tau Level as a Predictive Biomarker for Dementia and Related Endophenotypes. JAMA Neurol. 2019, 76, 598. [Google Scholar] [CrossRef]
- Mielke, M.M.; Hagen, C.E.; Xu, J.; Chai, X.; Vemuri, P.; Lowe, V.J.; Airey, D.C.; Knopman, D.S.; Roberts, R.O.; Machulda, M.M.; et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimer’s Dement. 2018, 14, 989–997. [Google Scholar] [CrossRef]
- Lim, C.Z.J.; Zhang, Y.; Chen, Y.; Zhao, H.; Stephenson, M.C.; Ho, N.R.Y.; Chen, Y.; Chung, J.; Reilhac, A.; Loh, T.P.; et al. Subtyping of circulating exosome-bound amyloid β reflects brain plaque deposition. Nat. Commun. 2019, 10, 1144. [Google Scholar] [CrossRef] [Green Version]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 26. [Google Scholar] [CrossRef]
- Yao, F.; Hong, X.; Li, S.; Zhang, Y.; Zhao, Q.; Du, W.; Wang, Y.; Ni, J. Urine-Based Biomarkers for Alzheimer’s Disease Identified Through Coupling Computational and Experimental Methods. J. Alzheimer’s Dis. 2018, 65, 421–431. [Google Scholar] [CrossRef]
- Watanabe, Y.; Hirao, Y.; Kasuga, K.; Tokutake, T.; Semizu, Y.; Kitamura, K.; Ikeuchi, T.; Nakamura, K.; Yamamoto, T. Molecular Network Analysis of the Urinary Proteome of Alzheimer’s Disease Patients. Dement. Geriatr. Cogn. Dis. Extra 2019, 9, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Orešič, M.; Hyötyläinen, T.; Herukka, S.-K.; Sysi-Aho, M.; Mattila, I.; Seppänan-Laakso, T.; Julkunen, V.; Gopalacharyulu, P.V.; Hallikainen, M.; Koikkalainen, J.; et al. Metabolome in progression to Alzheimer’s disease. Transl. Psychiatry 2011, 1, e57. [Google Scholar] [CrossRef]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef]
- Salvadores, N.; Shahnawaz, M.; Scarpini, E.; Tagliavini, F.; Soto, C. Detection of Misfolded Aβ Oligomers for Sensitive Biochemical Diagnosis of Alzheimer’s Disease. Cell Rep. 2014, 7, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Bongianni, M.; Ladogana, A.; Capaldi, S.; Klotz, S.; Baiardi, S.; Cagnin, A.; Perra, D.; Fiorini, M.; Poleggi, A.; Legname, G.; et al. α-Synuclein RT-QuIC assay in cerebrospinal fluid of patients with dementia with Lewy bodies. Ann. Clin. Transl. Neurol. 2019, 6, 2120–2126. [Google Scholar] [CrossRef]
- Rossi, M.; Candelise, N.; Baiardi, S.; Capellari, S.; Giannini, G.; Orrù, C.D.; Antelmi, E.; Mammana, A.; Hughson, A.G.; Calandra-Buonaura, G.; et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 2020, 140, 49–62. [Google Scholar] [CrossRef]
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.W.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Mendez, N.; Ishii, R.; Trenkwalder, C.; Mollenhauer, B.; Soto, C. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of α-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 2017, 74, 163. [Google Scholar] [CrossRef]
- Saijo, E.; Metrick, M.A.; Koga, S.; Parchi, P.; Litvan, I.; Spina, S.; Boxer, A.; Rojas, J.C.; Galasko, D.; Kraus, A.; et al. 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 2020, 139, 63–77. [Google Scholar] [CrossRef]
- Scialò, C.; Tran, T.H.; Salzano, G.; Novi, G.; Caponnetto, C.; Chiò, A.; Calvo, A.; Canosa, A.; Moda, F.; Caroppo, P.; et al. TDP-43 real time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020, 2, fcaa142. [Google Scholar] [CrossRef]
- Manne, S.; Kondru, N.; Hepker, M.; Jin, H.; Anantharam, V.; Lewis, M.; Huang, X.; Kanthasamy, A.; Kanthasamy, A.G. Ultrasensitive Detection of Aggregated α-Synuclein in Glial Cells, Human Cerebrospinal Fluid, and Brain Tissue Using the RT-QuIC Assay: New High-Throughput Neuroimmune Biomarker Assay for Parkinsonian Disorders. J. Neuroimmune Pharmacol. 2019, 14, 423–435. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.M.G.; Elia, A.E.; Portaleone, S.M.; Cazzaniga, F.A.; Rossi, M.; Bistaffa, E.; De Cecco, E.; Narkiewicz, J.; Salzano, G.; Carletta, O.; et al. Efficient RT-QuIC seeding activity for α-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Transl. Neurodegener. 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Saijo, E.; Ghetti, B.; Zanusso, G.; Oblak, A.; Furman, J.L.; Diamond, M.I.; Kraus, A.; Caughey, B. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017, 133, 751–765. [Google Scholar] [CrossRef]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Lawton, M.; Baig, F.; Rolinski, M.; Ruffman, C.; Nithi, K.; May, M.T.; Ben-Shlomo, Y.; Hu, M.T.M. Parkinson’s Disease Subtypes in the Oxford Parkinson Disease Centre (OPDC) Discovery Cohort. J. Parkinsons. Dis. 2015, 5, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Peelaerts, W.; Baekelandt, V. ɑ-Synuclein strains and the variable pathologies of synucleinopathies. J. Neurochem. 2016, 139, 256–274. [Google Scholar] [CrossRef] [Green Version]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Candelise, N.; Schmitz, M.; Llorens, F.; Villar-Piqué, A.; Cramm, M.; Thom, T.; Silva Correia, S.M.; Cunha, J.E.G.; Möbius, W.; Outeiro, T.F.; et al. Seeding variability of different alpha synuclein strains in synucleinopathies. Ann. Neurol. 2019, 85, 691–703. [Google Scholar] [CrossRef]
- Groveman, B.R.; Orrù, C.D.; Hughson, A.G.; Raymond, L.D.; Zanusso, G.; Ghetti, B.; Campbell, K.J.; Safar, J.; Galasko, D.; Caughey, B. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol. Commun. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Kang, U.J.; Boehme, A.K.; Fairfoul, G.; Shahnawaz, M.; Ma, T.C.; Hutten, S.J.; Green, A.; Soto, C. Comparative study of cerebrospinal fluid α-synuclein seeding aggregation assays for diagnosis of Parkinson’s disease. Mov. Disord. 2019, 34, 536–544. [Google Scholar] [CrossRef]
- Capitini, C.; Patel, J.R.; Natalello, A.; D’Andrea, C.; Relini, A.; Jarvis, J.A.; Birolo, L.; Peduzzo, A.; Vendruscolo, M.; Matteini, P.; et al. Structural differences between toxic and nontoxic HypF-N oligomers. Chem. Commun. 2018, 54, 8637–8640. [Google Scholar] [CrossRef]
- Kong, K.; Kendall, C.; Stone, N.; Notingher, I. Raman spectroscopy for medical diagnostics — From in-vitro biofluid assays to in-vivo cancer detection. Adv. Drug Deliv. Rev. 2015, 89, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krafft, C.; Popp, J. The many facets of Raman spectroscopy for biomedical analysis. Anal. Bioanal. Chem. 2015, 407, 699–717. [Google Scholar] [CrossRef] [PubMed]
- Banchelli, M.; de Angelis, M.; D’Andrea, C.; Pini, R.; Matteini, P. Triggering molecular assembly at the mesoscale for advanced Raman detection of proteins in liquid. Sci. Rep. 2018, 8, 1033. [Google Scholar] [CrossRef] [Green Version]
- Eravuchira, P.; Banchelli, M.; D’Andrea, C.; De Angelis, M.; Matteini, P.; Gannot, I. Hollow core photonic crystal fiber-assisted Raman spectroscopy as a tool for the detection of Alzheimer’s disease biomarkers. J. Biomed. Opt. 2020, 25, 1–10. [Google Scholar] [CrossRef]
- Banchelli, M.; Amicucci, C.; Ruggiero, E.; D’Andrea, C.; Cottat, M.; Ciofini, D.; Osticioli, I.; Ghini, G.; Siano, S.; Pini, R.; et al. Spot-on SERS Detection of Biomolecules with Laser-Patterned Dot Arrays of Assembled Silver Nanowires. ChemNanoMat 2019, 5, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, C.; Foti, A.; Cottat, M.; Banchelli, M.; Capitini, C.; Barreca, F.; Canale, C.; de Angelis, M.; Relini, A.; Maragò, O.M.; et al. Nanoscale Discrimination between Toxic and Nontoxic Protein Misfolded Oligomers with Tip-Enhanced Raman Spectroscopy. Small 2018, 14, 1800890. [Google Scholar] [CrossRef]
- Matteini, P.; de Angelis, M.; Ulivi, L.; Centi, S.; Pini, R. Concave gold nanocube assemblies as nanotraps for surface-enhanced Raman scattering-based detection of proteins. Nanoscale 2015, 7, 3474–3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, L.; Arenal, R.; Mannini, B.; Chiti, F.; Pini, R.; Matteini, P.; Alvarez-Puebla, R.A. SERS Detection of Amyloid Oligomers on Metallorganic-Decorated Plasmonic Beads. ACS Appl. Mater. Interfaces 2015, 7, 9420–9428. [Google Scholar] [CrossRef]
- Matteini, P.; Cottat, M.; Tavanti, F.; Panfilova, E.; Scuderi, M.; Nicotra, G.; Menziani, M.C.; Khlebtsov, N.; de Angelis, M.; Pini, R. Site-Selective Surface-Enhanced Raman Detection of Proteins. ACS Nano 2017, 11, 918–926. [Google Scholar] [CrossRef]
- Banchelli, M.; Cascella, R.; D’Andrea, C.; Cabaj, L.; Osticioli, I.; Ciofini, D.; Li, M.S.; Skupień, K.; de Angelis, M.; Siano, S.; et al. Nanoscopic insights into the surface conformation of neurotoxic amyloid β oligomers. RSC Adv. 2020, 10, 21907–21913. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bistaffa, E.; Tagliavini, F.; Matteini, P.; Moda, F. Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease. Brain Sci. 2020, 10, 815. https://doi.org/10.3390/brainsci10110815
Bistaffa E, Tagliavini F, Matteini P, Moda F. Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease. Brain Sciences. 2020; 10(11):815. https://doi.org/10.3390/brainsci10110815
Chicago/Turabian StyleBistaffa, Edoardo, Fabrizio Tagliavini, Paolo Matteini, and Fabio Moda. 2020. "Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease" Brain Sciences 10, no. 11: 815. https://doi.org/10.3390/brainsci10110815
APA StyleBistaffa, E., Tagliavini, F., Matteini, P., & Moda, F. (2020). Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease. Brain Sciences, 10(11), 815. https://doi.org/10.3390/brainsci10110815