Caenorhabditis elegans Models to Investigate the Mechanisms Underlying Tau Toxicity in Tauopathies



Abstract
:1. Introduction
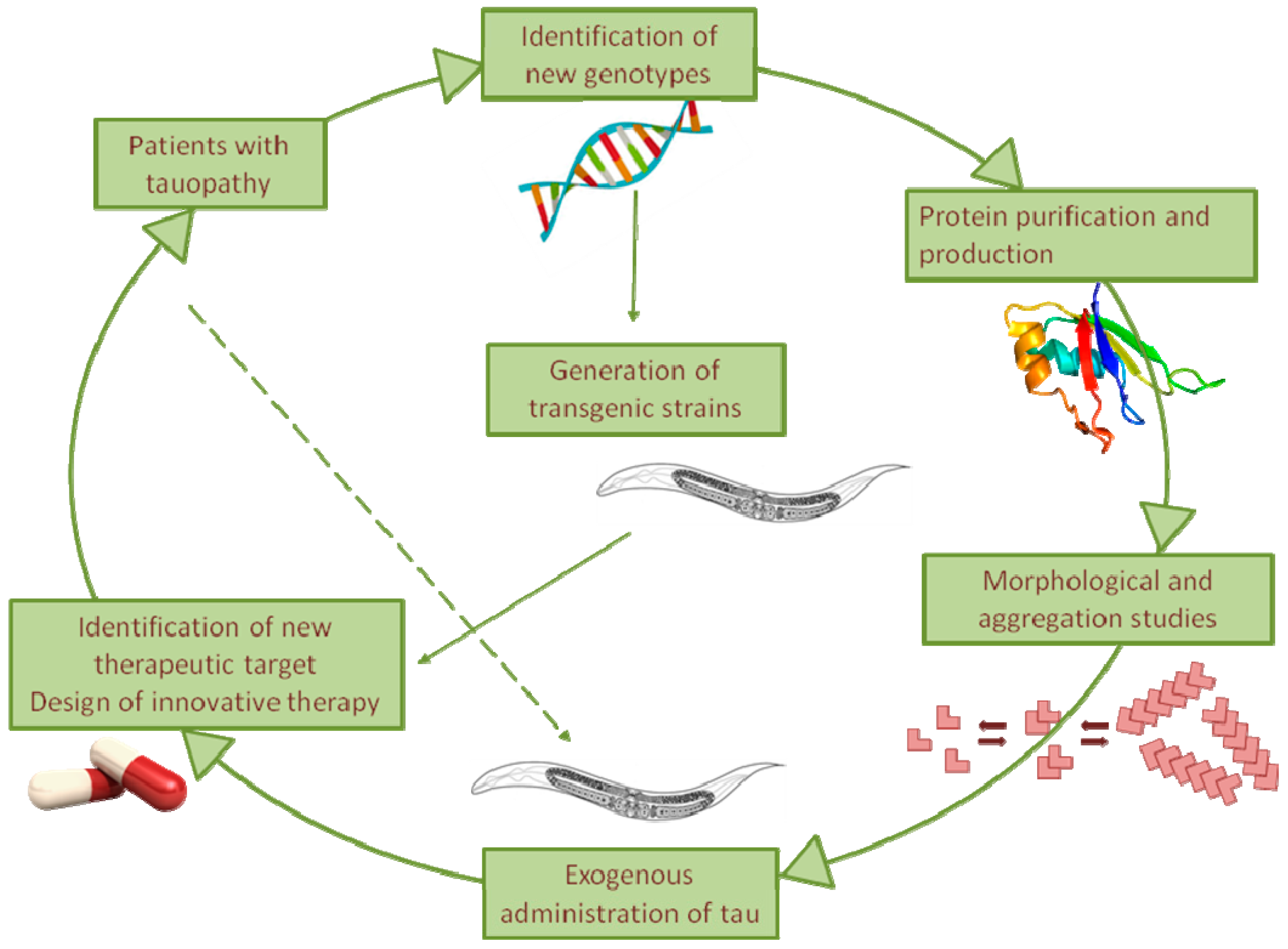
2. Transgenic C. elegans Models of Tauopathy
3. Other Non-Mammalian Models of Tauopathy
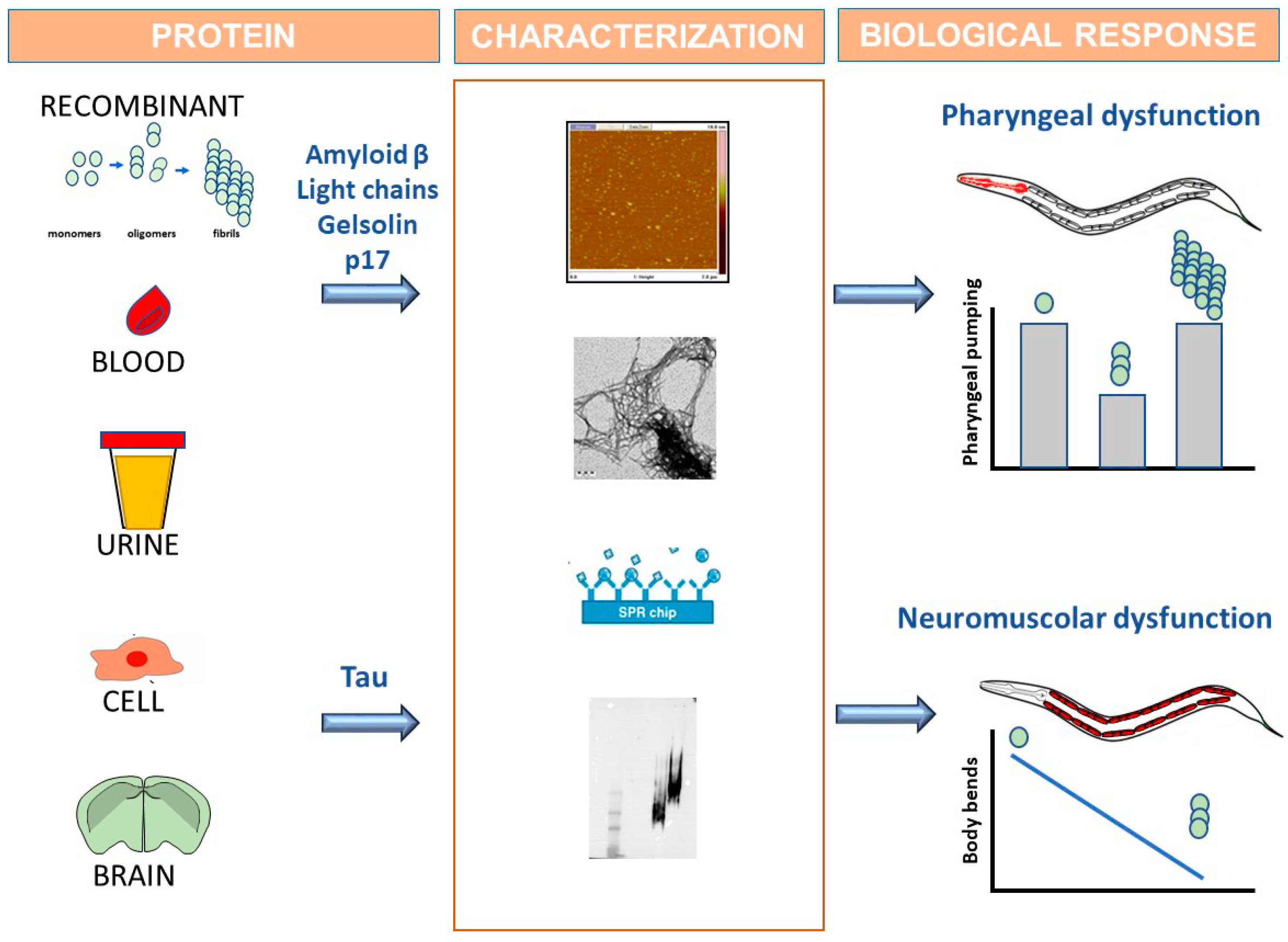
4. C. elegans Recognizes the Toxic Component of Tau
5. C. elegans for Drug Discovery
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haass, C.; Neumann, M. Frontotemporal dementia: From molecular mechanisms to therapy. J. Neurochem. 2016, 138 (Suppl. 1), 3–5. [Google Scholar] [CrossRef] [PubMed]
- Pollock, N.J.; Mirra, S.S.; Binder, L.I.; Hansen, L.A.; Wood, J.G. Filamentous aggregates in Pick’s disease, progressive supranuclear palsy, and Alzheimer’s disease share antigenic determinants with microtubule-associated protein, tau. Lancet 1986, 2, 1211. [Google Scholar] [CrossRef]
- Lee, G.; Leugers, C.J. Tau and tauopathies. Prog. Mol. Biol. Transl. Sci. 2012, 107, 263–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Freund, R.K.; Gibson, E.S.; Potter, H.; Dell’Acqua, M.L. Inhibition of the Motor Protein Eg5/Kinesin-5 in Amyloid β-Mediated Impairment of Hippocampal Long-Term Potentiation and Dendritic Spine Loss. Mol. Pharmacol. 2016, 89, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M. Tau protein and neurodegeneration. Semin. Cell Dev. Biol. 2004, 15, 45–49. [Google Scholar] [CrossRef]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G. Tau gene mutations and neurodegeneration. Biochem. Soc. Symp. 2001, 67, 59–71. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Crowther, R.A.; Chen, S.G.; Parchi, P.; Tabaton, M.; Lanska, D.J.; Markesbery, W.R.; Wilhelmsen, K.C.; Dickson, D.W.; et al. Tau gene mutation in familial progressive subcortical gliosis. Nat. Med. 1999, 5, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Smith, M.J.; Goedert, M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998, 437, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Nacharaju, P.; Lewis, J.; Easson, C.; Yen, S.; Hackett, J.; Hutton, M.; Yen, S.H. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999, 447, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 2010, 49, 10039–10041. [Google Scholar] [CrossRef]
- Ayers, J.I.; Giasson, B.I.; Borchelt, D.R. Prion-like Spreading in Tauopathies. Biol. Psychiatry 2018, 83, 337–346. [Google Scholar] [CrossRef]
- Fuster-Matanzo, A.; Hernández, F.; Ávila, J. Tau Spreading Mechanisms; Implications for Dysfunctional Tauopathies. Int. J. Mol. Sci. 2018, 19, 645. [Google Scholar] [CrossRef] [Green Version]
- Hobert, O. The neuronal genome of Caenorhabditis elegans. In WormBook; Columbia University Medical Center, HHMI: New York, NY, USA, 2013; pp. 1–106. [Google Scholar] [CrossRef]
- Girard, L.R.; Fiedler, T.J.; Harris, T.W.; Carvalho, F.; Antoshechkin, I.; Han, M.; Sternberg, P.W.; Stein, L.D.; Chalfie, M. WormBook: The online review of Caenorhabditis elegans biology. Nucleic Acids Res. 2007, 35, D472–D475. [Google Scholar] [CrossRef] [PubMed]
- Teschendorf, D.; Link, C.D. What have worm models told us about the mechanisms of neuronal dysfunction in human neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Sandhof, C.A.; Hoppe, S.O.; Tittelmeier, J.; Nussbaum-Krammer, C.C. elegans Models to Study the Propagation of Prions and Prion-Like Proteins. Biomolecules 2020, 10, 1188. [Google Scholar] [CrossRef]
- Link, C.D. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum-Krammer, C.I.; Morimoto, R.I. Caenorhabditis elegans as a model system for studying non-cell-autonomous mechanisms in protein-misfolding diseases. Dis. Model. Mech. 2014, 7, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Culetto, E.; Sattelle, D.B. A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum. Mol. Genet. 2000, 9, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Brandt, R.; Gergou, A.; Wacker, I.; Fath, T.; Hutter, H. A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 2009, 30, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef] [Green Version]
- Miyasaka, T.; Ding, Z.; Gengyo-Ando, K.; Oue, M.; Yamaguchi, H.; Mitani, S.; Ihara, Y. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol. Dis. 2005, 20, 372–383. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Mandelkow, E.; Mandelkow, E.-M. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener. 2016, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Butler, V.J.; Salazar, D.A.; Soriano-Castell, D.; Alves-Ferreira, M.; Dennissen, F.J.A.; Vohra, M.; Oses-Prieto, J.A.; Li, K.H.; Wang, A.L.; Jing, B.; et al. Tau/MAPT disease-associated variant A152T alters tau function and toxicity via impaired retrograde axonal transport. Hum. Mol. Genet. 2019, 28, 1498–1514. [Google Scholar] [CrossRef] [Green Version]
- Sulston, J.E.; Horvitz, H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Sulston, J.E. Neuronal cell lineages in the nematode Caenorhabditis elegans. Cold Spring Harb. Symp. Quant. Biol. 1983, 48 Pt 2, 443–452. [Google Scholar] [CrossRef]
- Consortium*, T.C. elegans S. Genome Sequence of the Nematode C. elegans: A Platform for Investigating Biology. Science 1998, 282, 2012–2018. [Google Scholar] [CrossRef]
- Lai, C.H.; Chou, C.Y.; Ch’ang, L.Y.; Liu, C.S.; Lin, W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 2000, 10, 703–713. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Underwood, R.S.; Greenwald, I.; Shaye, D.D. OrthoList 2: A New Comparative Genomic Analysis of Human and Caenorhabditis elegans Genes. Genetics 2018, 210, 445–461. [Google Scholar] [CrossRef] [Green Version]
- Dimitriadi, M.; Hart, A.C. Neurodegenerative disorders: Insights from the nematode Caenorhabditis elegans. Neurobiol. Dis. 2010, 40, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Nollen, E.A.A.; Garcia, S.M.; van Haaften, G.; Kim, S.; Chavez, A.; Morimoto, R.I.; Plasterk, R.H.A. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2004, 101, 6403–6408. [Google Scholar] [CrossRef] [Green Version]
- Lejeune, F.-X.; Mesrob, L.; Parmentier, F.; Bicep, C.; Vazquez-Manrique, R.P.; Parker, J.A.; Vert, J.-P.; Tourette, C.; Neri, C. Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC Genom. 2012, 13, 91. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.A.; Selak, M.A.; Xiang, Z.; Krainc, D.; Neve, R.L.; Kraemer, B.C.; Watts, J.L.; Kalb, R.G. Reduced Activity of AMP-Activated Protein Kinase Protects against Genetic Models of Motor Neuron Disease. J. Neurosci. 2012, 32, 1123–1141. [Google Scholar] [CrossRef] [Green Version]
- van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.W.; Plasterk, R.H.A.; Nollen, E.A.A. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008, 4, e1000027. [Google Scholar] [CrossRef]
- Hsu, A.-L.; Murphy, C.T.; Kenyon, C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 2003, 300, 1142–1145. [Google Scholar] [CrossRef] [Green Version]
- Morley, J.F.; Morimoto, R.I. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 2004, 15, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaletta, T.; Hengartner, M.O. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–399. [Google Scholar] [CrossRef]
- Calamini, B.; Silva, M.C.; Madoux, F.; Hutt, D.M.; Khanna, S.; Chalfant, M.A.; Saldanha, S.A.; Hodder, P.; Tait, B.D.; Garza, D.; et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 2011, 8, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Fatouros, C.; Pir, G.J.; Biernat, J.; Koushika, S.P.; Mandelkow, E.; Mandelkow, E.-M.; Schmidt, E.; Baumeister, R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 2012, 21, 3587–3603. [Google Scholar] [CrossRef] [Green Version]
- Guthrie, C.R.; Schellenberg, G.D.; Kraemer, B.C. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet. 2009, 18, 1825–1838. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, B.; Schellenberg, G.D. Using Caenorhabditis elegans models of neurodegenerative disease to identify neuroprotective strategies. Int. Rev. Neurobiol. 2007, 77, 219–246. [Google Scholar] [CrossRef]
- Goedert, M.; Baur, C.P.; Ahringer, J.; Jakes, R.; Hasegawa, M.; Spillantini, M.G.; Smith, M.J.; Hill, F. PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J. Cell. Sci. 1996, 109 Pt 11, 2661–2672. [Google Scholar]
- Gordon, P.; Hingula, L.; Krasny, M.L.; Swienckowski, J.L.; Pokrywka, N.J.; Raley-Susman, K.M. The invertebrate microtubule-associated protein PTL-1 functions in mechanosensation and development in Caenorhabditis elegans. Dev. Genes Evol. 2008, 218, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Hashi, Y.; Kotani, S.; Adachi, T. A nematode microtubule-associated protein, PTL-1, closely resembles its mammalian counterparts in overall molecular architecture. Biosci. Biotechnol. Biochem. 2016, 80, 1107–1113. [Google Scholar] [CrossRef] [Green Version]
- McDermott, J.B.; Aamodt, S.; Aamodt, E. ptl-1, a Caenorhabditis elegans gene whose products are homologous to the tau microtubule-associated proteins. Biochemistry 1996, 35, 9415–9423. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diomede, L.; Di Fede, G.; Romeo, M.; Bagnati, R.; Ghidoni, R.; Fiordaliso, F.; Salio, M.; Rossi, A.; Catania, M.; Paterlini, A.; et al. Expression of A2V-mutated Aβ in Caenorhabditis elegans results in oligomer formation and toxicity. Neurobiol. Dis. 2014, 62, 521–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyasaka, T.; Xie, C.; Yoshimura, S.; Shinzaki, Y.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Ihara, Y. Curcumin improves tau-induced neuronal dysfunction of nematodes. Neurobiol. Aging 2016, 39, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Morelli, F.; Romeo, M.; Barzago, M.M.; Bolis, M.; Mattioni, D.; Rossi, G.; Tagliavini, F.; Bastone, A.; Salmona, M.; Diomede, L. V363I and V363A mutated tau affect aggregation and neuronal dysfunction differently in C. elegans. Neurobiol. Dis. 2018, 117, 226–234. [Google Scholar] [CrossRef]
- Rossi, G.; Bastone, A.; Piccoli, E.; Morbin, M.; Mazzoleni, G.; Fugnanesi, V.; Beeg, M.; Del Favero, E.; Cantù, L.; Motta, S.; et al. Different mutations at V363 MAPT codon are associated with atypical clinical phenotypes and show unusual structural and functional features. Neurobiol. Aging 2014, 35, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, M.; Bernardi, L.; Gallo, M.; Geracitano, S.; Colao, R.; Puccio, G.; Curcio, S.A.M.; Frangipane, F.; Mirabelli, M.; Tomaino, C.; et al. MAPT V363I variation in a sporadic case of frontotemporal dementia: Variable penetrant mutation or rare polymorphism? Alzheimer Dis. Assoc. Disord. 2011, 25, 96–99. [Google Scholar] [CrossRef]
- Bessi, V.; Bagnoli, S.; Nacmias, B.; Tedde, A.; Sorbi, S.; Bracco, L. Semantic dementia associated with mutation V363I in the tau gene. J. Neurol. Sci. 2010, 296, 112–114. [Google Scholar] [CrossRef]
- Munoz, D.G.; Ros, R.; Fatas, M.; Bermejo, F.; de Yebenes, J.G. Progressive nonfluent aphasia associated with a new mutation V363I in tau gene. Am. J. Alzheimers Dis. Dement. 2007, 22, 294–299. [Google Scholar] [CrossRef]
- Fortini, M.E.; Bonini, N.M. Modeling human neurodegenerative diseases in Drosophila: On a wing and a prayer. Trends Genet. 2000, 16, 161–167. [Google Scholar] [CrossRef]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A Systematic Analysis of Human Disease-Associated Gene Sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidary, G.; Fortini, M.E. Identification and characterization of the Drosophila tau homolog. Mech. Dev. 2001, 108, 171–178. [Google Scholar] [CrossRef]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.-K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human Wild-Type Tau Interacts with wingless Pathway Components and Produces Neurofibrillary Pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration Without Neurofibrillary Tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, I.; Yang, Y.; Lu, B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 2004, 116, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Chee, F.C.; Mudher, A.; Cuttle, M.F.; Newman, T.A.; MacKay, D.; Lovestone, S.; Shepherd, D. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 2005, 20, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Shepherd, D.; Newman, T.A.; Mildren, P.; Jukes, J.P.; Squire, A.; Mears, A.; Drummond, J.A.; Berg, S.; MacKay, D.; et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 2004, 9, 522–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mershin, A.; Pavlopoulos, E.; Fitch, O.; Braden, B.C.; Nanopoulos, D.V.; Skoulakis, E.M.C. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn. Mem. 2004, 11, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Khurana, V. Modeling Tauopathy in the fruit fly Drosophila melanogaster. J. Alzheimers Dis. 2008, 15, 541–553. [Google Scholar] [CrossRef]
- Shulman, J.M.; Feany, M.B. Genetic modifiers of tauopathy in Drosophila. Genetics 2003, 165, 1233–1242. [Google Scholar]
- Steinhilb, M.L.; Dias-Santagata, D.; Mulkearns, E.E.; Shulman, J.M.; Biernat, J.; Mandelkow, E.-M.; Feany, M.B. S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J. Neurosci. Res. 2007, 85, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Rupp, B.; Wullimann, M.F.; Reichert, H. The zebrafish brain: A neuroanatomical comparison with the goldfish. Anat. Embryol. 1996, 194, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Wullimann, M.F. An evolutionary interpretation of teleostean forebrain anatomy. Brain Behav. Evol. 2009, 74, 30–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rink, E.; Wullimann, M.F. Connections of the ventral telencephalon (subpallium) in the zebrafish (Danio rerio). Brain Res. 2004, 1011, 206–220. [Google Scholar] [CrossRef]
- Mueller, T.; Vernier, P.; Wullimann, M.F. The adult central nervous cholinergic system of a neurogenetic model animal, the zebrafish Danio rerio. Brain Res. 2004, 1011, 156–169. [Google Scholar] [CrossRef]
- Sallinen, V.; Torkko, V.; Sundvik, M.; Reenilä, I.; Khrustalyov, D.; Kaslin, J.; Panula, P. MPTP and MPP+ target specific aminergic cell populations in larval zebrafish. J. Neurochem. 2009, 108, 719–731. [Google Scholar] [CrossRef]
- Meng, S.; Ryu, S.; Zhao, B.; Zhang, D.-Q.; Driever, W.; McMahon, D.G. Targeting retinal dopaminergic neurons in tyrosine hydroxylase-driven green fluorescent protein transgenic zebrafish. Mol. Vis. 2008, 14, 2475–2483. [Google Scholar]
- Bai, Q.; Burton, E.A. Cis-acting elements responsible for dopaminergic neuron-specific expression of zebrafish slc6a3 (dopamine transporter) in vivo are located remote from the transcriptional start site. Neuroscience 2009, 164, 1138–1151. [Google Scholar] [CrossRef]
- Anichtchik, O.V.; Kaslin, J.; Peitsaro, N.; Scheinin, M.; Panula, P. Neurochemical and behavioural changes in zebrafish Danio rerio after systemic administration of 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Neurochem. 2004, 88, 443–453. [Google Scholar] [CrossRef]
- Tomasiewicz, H.G.; Flaherty, D.B.; Soria, J.P.; Wood, J.G. Transgenic zebrafish model of neurodegeneration. J. Neurosci. Res. 2002, 70, 734–745. [Google Scholar] [CrossRef]
- Jicha, G.A.; Bowser, R.; Kazam, I.G.; Davies, P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J. Neurosci. Res. 1997, 48, 128–132. [Google Scholar] [CrossRef]
- Gao, Y.; Li, P.; Li, L. Transgenic zebrafish that express tyrosine hydroxylase promoter in inner retinal cells. Dev. Dyn. 2005, 233, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Lei, L.; Lai, C.; Tang, Z. Tau Protein and Zebrafish Models for Tau-Induced Neurodegeneration. J. Alzheimers Dis. 2019, 69, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Bretaud, S.; Allen, C.; Ingham, P.W.; Bandmann, O. p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. J. Neurochem. 2007, 100, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Mullett, S.J.; Garver, J.A.; Hinkle, D.A.; Burton, E.A. Zebrafish DJ-1 is evolutionarily conserved and expressed in dopaminergic neurons. Brain Res. 2006, 1113, 33–44. [Google Scholar] [CrossRef]
- Anichtchik, O.; Diekmann, H.; Fleming, A.; Roach, A.; Goldsmith, P.; Rubinsztein, D.C. Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J. Neurosci. 2008, 28, 8199–8207. [Google Scholar] [CrossRef]
- Sun, Z.; Gitler, A.D. Discovery and characterization of three novel synuclein genes in zebrafish. Dev. Dyn. 2008, 237, 2490–2495. [Google Scholar] [CrossRef]
- Flinn, L.; Mortiboys, H.; Volkmann, K.; Köster, R.W.; Ingham, P.W.; Bandmann, O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 2009, 132, 1613–1623. [Google Scholar] [CrossRef] [Green Version]
- Campbell, W.A.; Yang, H.; Zetterberg, H.; Baulac, S.; Sears, J.A.; Liu, T.; Wong, S.T.C.; Zhong, T.P.; Xia, W. Zebrafish lacking Alzheimer presenilin enhancer 2 (Pen-2) demonstrate excessive p53-dependent apoptosis and neuronal loss. J. Neurochem. 2006, 96, 1423–1440. [Google Scholar] [CrossRef]
- Groth, C.; Nornes, S.; McCarty, R.; Tamme, R.; Lardelli, M. Identification of a second presenilin gene in zebrafish with similarity to the human Alzheimer’s disease gene presenilin2. Dev. Genes Evol. 2002, 212, 486–490. [Google Scholar] [CrossRef]
- Leimer, U.; Lun, K.; Romig, H.; Walter, J.; Grünberg, J.; Brand, M.; Haass, C. Zebrafish (Danio rerio) presenilin promotes aberrant amyloid beta-peptide production and requires a critical aspartate residue for its function in amyloidogenesis. Biochemistry 1999, 38, 13602–13609. [Google Scholar] [CrossRef] [PubMed]
- Long, Q.; Meng, A.; Wang, H.; Jessen, J.R.; Farrell, M.J.; Lin, S. GATA-1 expression pattern can be recapitulated in living transgenic zebrafish using GFP reporter gene. Development 1997, 124, 4105–4111. [Google Scholar] [PubMed]
- Meng, A.; Tang, H.; Ong, B.A.; Farrell, M.J.; Lin, S. Promoter analysis in living zebrafish embryos identifies a cis-acting motif required for neuronal expression of GATA-2. Proc. Natl. Acad. Sci. USA 1997, 94, 6267–6272. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Ueshima, E.; Muraoka, O.; Tanaka, H.; Yeo, S.Y.; Huh, T.L.; Miki, N. Zebrafish elav/HuC homologue as a very early neuronal marker. Neurosci. Lett. 1996, 216, 109–112. [Google Scholar] [CrossRef]
- Chen, M.; Martins, R.N.; Lardelli, M. Complex splicing and neural expression of duplicated tau genes in zebrafish embryos. J. Alzheimers Dis. 2009, 18, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.-M.; Berg, S.; Hellberg, S.; Fälting, J.; Distel, M.; Köster, R.W.; Schmid, B.; et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef] [Green Version]
- Bai, Q.; Garver, J.A.; Hukriede, N.A.; Burton, E.A. Generation of a transgenic zebrafish model of Tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res. 2007, 35, 6501–6516. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.; Lee, S.E.; Wojta, K.; Ramos, E.M.; Klein, E.; Chen, J.; Boxer, A.L.; Gorno-Tempini, M.L.; Geschwind, D.H.; Schlotawa, L.; et al. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 2017, 140, 1128–1146. [Google Scholar] [CrossRef]
- Beeg, M.; Diomede, L.; Stravalaci, M.; Salmona, M.; Gobbi, M. Novel approaches for studying amyloidogenic peptides/proteins. Curr. Opin. Pharmacol. 2013, 13, 797–801. [Google Scholar] [CrossRef]
- Mishra, S.; Guan, J.; Plovie, E.; Seldin, D.C.; Connors, L.H.; Merlini, G.; Falk, R.H.; MacRae, C.A.; Liao, R. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H95–H103. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Joshi, S.; Ward, J.E.; Buys, E.P.; Mishra, D.; Mishra, D.; Morgado, I.; Fisch, S.; Lavatelli, F.; Merlini, G.; et al. Zebrafish model of amyloid light chain cardiotoxicity: Regeneration versus degeneration. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1158–H1166. [Google Scholar] [CrossRef] [PubMed]
- Stravalaci, M.; Tapella, L.; Beeg, M.; Rossi, A.; Joshi, P.; Pizzi, E.; Mazzanti, M.; Balducci, C.; Forloni, G.; Biasini, E.; et al. The Anti-Prion Antibody 15B3 Detects Toxic Amyloid-β Oligomers. J. Alzheimers Dis. 2016, 53, 1485–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeinolabediny, Y.; Caccuri, F.; Colombo, L.; Morelli, F.; Romeo, M.; Rossi, A.; Schiarea, S.; Ciaramelli, C.; Airoldi, C.; Weston, R.; et al. HIV-1 matrix protein p17 misfolding forms toxic amyloidogenic assemblies that induce neurocognitive disorders. Sci. Rep. 2017, 7, 10313. [Google Scholar] [CrossRef] [PubMed]
- Diomede, L.; Rognoni, P.; Lavatelli, F.; Romeo, M.; del Favero, E.; Cantù, L.; Ghibaudi, E.; di Fonzo, A.; Corbelli, A.; Fiordaliso, F.; et al. A Caenorhabditis elegans-based assay recognizes immunoglobulin light chains causing heart amyloidosis. Blood 2014, 123, 3543–3552. [Google Scholar] [CrossRef] [Green Version]
- Diomede, L.; Romeo, M.; Rognoni, P.; Beeg, M.; Foray, C.; Ghibaudi, E.; Palladini, G.; Cherny, R.A.; Verga, L.; Capello, G.L.; et al. Cardiac Light Chain Amyloidosis: The Role of Metal Ions in Oxidative Stress and Mitochondrial Damage. Antioxid. Redox Signal. 2017, 27, 567–582. [Google Scholar] [CrossRef]
- Giorgino, T.; Mattioni, D.; Hassan, A.; Milani, M.; Mastrangelo, E.; Barbiroli, A.; Verhelle, A.; Gettemans, J.; Barzago, M.M.; Diomede, L.; et al. Nanobody interaction unveils structure, dynamics and proteotoxicity of the Finnish-type amyloidogenic gelsolin variant. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 648–660. [Google Scholar] [CrossRef]
- Stravalaci, M.; Bastone, A.; Beeg, M.; Cagnotto, A.; Colombo, L.; Di Fede, G.; Tagliavini, F.; Cantù, L.; Del Favero, E.; Mazzanti, M.; et al. Specific recognition of biologically active amyloid-β oligomers by a new surface plasmon resonance-based immunoassay and an in vivo assay in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 27796–27805. [Google Scholar] [CrossRef] [Green Version]
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Primers 2018, 4, 38. [Google Scholar] [CrossRef]
- Zanier, E.R.; Barzago, M.M.; Vegliante, G.; Romeo, M.; Bertani, I.; Natale, C.; Colnaghi, L.; Colombo, L.; Russo, L.; Micotti, E.; et al. Role of misfolded tau in the onset and progression of brain toxicity after trauma. bioRxiv 2020, 2020, 159301. [Google Scholar] [CrossRef]
- Wechalekar, A.D.; Whelan, C. Encouraging impact of doxycycline on early mortality in cardiac light chain (AL) amyloidosis. Blood Cancer J. 2017, 7, e546. [Google Scholar] [CrossRef] [Green Version]
- Fluharty, B.R.; Biasini, E.; Stravalaci, M.; Sclip, A.; Diomede, L.; Balducci, C.; La Vitola, P.; Messa, M.; Colombo, L.; Forloni, G.; et al. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 2013, 288, 7857–7866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artal-Sanz, M.; de Jong, L.; Tavernarakis, N. Caenorhabditis elegans: A versatile platform for drug discovery. Biotechnol. J. 2006, 1, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ebert, P.R. 5-Methoxyindole-2-carboxylic acid (MICA) suppresses Aβ-mediated pathology in C. elegans. Exp. Gerontol. 2018, 108, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, W. Suppression of the dihydrolipoamide dehydrogenase gene (dld-1) protects against the toxicity of human amyloid beta in C. elegans model of Alzheimer’s disease. bioRxiv 2017, 228429. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, W. Dihydrolipoamide dehydrogenase suppression induces human tau phosphorylation by increasing whole body glucose levels in a C. elegans model of Alzheimer’s Disease. Exp. Brain Res. 2018, 236, 2857–2866. [Google Scholar] [CrossRef]
- Jovanov-Milošević, N.; Petrović, D.; Sedmak, G.; Vukšić, M.; Hof, P.R.; Simić, G. Human fetal tau protein isoform: Possibilities for Alzheimer’s disease treatment. Int. J. Biochem. Cell Biol. 2012, 44, 1290–1294. [Google Scholar] [CrossRef] [Green Version]
- Gamir-Morralla, A.; Sacristán, S.; Medina, M.; Iglesias, T. Effects of Thioflavin T and GSK-3 Inhibition on Lifespan and Motility in a Caenorhabditis elegans Model of Tauopathy. J. Alzheimers Dis. Rep. 2019, 3, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Stoilova, T.; Colombo, L.; Forloni, G.; Tagliavini, F.; Salmona, M. A new face for old antibiotics: Tetracyclines in treatment of amyloidoses. J. Med. Chem. 2013, 56, 5987–6006. [Google Scholar] [CrossRef]
- Beeg, M.; Stravalaci, M.; Romeo, M.; Carrá, A.D.; Cagnotto, A.; Rossi, A.; Diomede, L.; Salmona, M.; Gobbi, M. Clusterin Binds to Aβ1-42 Oligomers with High Affinity and Interferes with Peptide Aggregation by Inhibiting Primary and Secondary Nucleation. J. Biol. Chem. 2016, 291, 6958–6966. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Promoter::Transgene | Expression Pattern | Phenotype | Reference |
|---|---|---|---|
| Paex-3::tau WT Paex-3::tau V337M Paex-3::tau P301L | Pan-neuronal | Uncoordinated movement Nerve cord degeneration Insoluble tau accumulation | [26] |
| Pmec-7::tau WT Pmec-7::tau P301L Pmec-7::tau R406W | Mechanosensory neurons | Decrease in touch response Neuritic abnormalities and microtubular loss Tau accumulation | [27] |
| Prgef-1::tauWT Prfef-1::tau PHP ° | Pan-neuronal | Uncoordinated locomotion Defect in motor neuronal development | [25] |
| Prab-3::tau F3ΔK280 Prab-3::tauF3ΔK280-PP * | Pan-neuronal | Locomotion impairment Motor neuron damage Tau aggregation | [46] |
| Punc-119::tau WT Punc-119::tau R406W | Pan-neuronal | Uncoordinated movement Neuritic abnormalities and microtubular loss | [55] |
| Psnb-1::tauWT Psnb-1::tauA152T | Pan-neuronal | Locomotion impairment Paralysis Neuronal dysfunction | [28] |
| Paex-3::tauWT Paex-3::tauV363A Paex-3::tauV363I | Pan-neuronal | Locomotion impairment Pharyngeal dysfunction Insoluble tau accumulation Synaptic impairment | [56] |
| Paex-3::tauWT Paex-3::tauA152T Paex-3::tauA152E | Pan-neuronal | Developmental toxicity Impaired retrograde axonal transport | [29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natale, C.; Barzago, M.M.; Diomede, L. Caenorhabditis elegans Models to Investigate the Mechanisms Underlying Tau Toxicity in Tauopathies. Brain Sci. 2020, 10, 838. https://doi.org/10.3390/brainsci10110838
Natale C, Barzago MM, Diomede L. Caenorhabditis elegans Models to Investigate the Mechanisms Underlying Tau Toxicity in Tauopathies. Brain Sciences. 2020; 10(11):838. https://doi.org/10.3390/brainsci10110838
Chicago/Turabian StyleNatale, Carmina, Maria Monica Barzago, and Luisa Diomede. 2020. "Caenorhabditis elegans Models to Investigate the Mechanisms Underlying Tau Toxicity in Tauopathies" Brain Sciences 10, no. 11: 838. https://doi.org/10.3390/brainsci10110838
APA StyleNatale, C., Barzago, M. M., & Diomede, L. (2020). Caenorhabditis elegans Models to Investigate the Mechanisms Underlying Tau Toxicity in Tauopathies. Brain Sciences, 10(11), 838. https://doi.org/10.3390/brainsci10110838