1. Introduction
Research on brain aging and on neurodegenerative diseases is one of the most important challenges that the scientific community has been facing in recent years. The mechanisms underlying brain aging are many and closely related. Recently, an important role has been demonstrated for a class of cell growth and survival factors called neurotrophins [
1]. Neurotrophins belong to a family of small proteins playing a fundamental role in both the central and the peripheral nervous systems. The main functions of these proteins are the regulation of axonal growth and neuronal differentiation. The neurotrophins family includes nerve growth factor, neurotrophin-3, neurotrophin-4/5 and, even more important, brain-derived neurotrophic factor (BDNF) [
2]. BDNF is the most abundant neurotrophin in the brain and appears essential for neuronal survival during the development and formation of neural networks in the adult brain, exerting its biological actions through tyrosine receptor kinase B (TrkB receptors) [
3]. It is produced in the brain by different types of cells and can be transported outside the brain through the blood–brain barrier (BBB). BDNF plays a fundamental role during brain development as it supports the survival and differentiation of neuronal populations of the peripheral and central nervous systems and regulates synaptogenesis, synaptic transmission and plasticity. Furthermore, BDNF plays a crucial role in learning and memory mechanisms [
4]. BDNF and other neurotrophins are now considered to be growth factors with a wide spectrum of functions also outside the nervous system, including the modulation and regulation of the immune function [
5]. Moreover, BDNF and its specific precursors may have a role in sustaining thymocyte precursor survival and supporting the thymocyte differentiation process [
6].
A preclinical study stated that the dysregulation of BDNF signaling is involved in several neurodegenerative disorders, including Alzheimer’s disease, and leads to a deficit in age-related learning [
7]. BDNF has also been shown to be able to interact with oxygen radicals (ROS) whose imbalance is involved in the mechanisms of aging, neurodegenerative diseases and some neuropsychiatric disorders [
7]. In brain aging there is a decline in the normal antioxidant potential, which leads to an increase in the brain’s vulnerability to the harmful effects of oxidative damage [
8]. BDNF is considered one of the protective agents against oxidative stress in the central nervous system [
9]. Some brain areas particularly involved in neurodegenerative diseases such as the hippocampus, the
substantia nigra, the amygdala and the frontal cortex are particularly sensitive to oxidative stress [
10]. The role of ROS as a marker of brain aging is supported by numerous studies demonstrating that an increase in these substances is related to the reduction of mitochondrial function [
11,
12]. Several clinical studies have shown changes in blood concentration of BDNF in patients with neuropsychiatric disorders such as major depression [
13], schizophrenia [
14] and Alzheimer’s disease [
15]. Studies conducted both in vitro and in vivo have shown that the expression of BDNF and of its specific TrkB receptor is essential to keep an appropriate number of proliferating stem cells, for the differentiation of neuronal populations and for the maturation of excitatory synapses [
2].
The possibility of using exogenous BDNF as a therapeutic approach against neurodegenerative diseases has been hypothesized in recent years. However, supplementation of exogenous BDNF presents several problems. The main one depends on the amount of BDNF that reaches the brain. If such amount is too low, it might not be enough to produce the desirable effects. On the other hand, if it is too high it might paradoxically be dangerous, as it might cause, for example, TrkB receptors to be downregulated, thus reducing the intracellular machinery linked to BDNF. The possibility that BDNF can cross the BBB is rather controversial. While some authors argue that it is not clear whether BDNF can readily cross the BBB [
16], others indicate that BDNF is able to [
17]. In an attempt to overcome such delivery problems, different methods have been developed, which, however, did not thoroughly solve the problem [
18]. Other studies have reported further problems related to the administration of neurotrophins in humans, depending on the dose and pharmacokinetics of these molecules [
19]. A promising way to achieve a fine regulation of physiological mechanisms could be the use of low-dose substances, as demonstrated by numerous studies available in the literature [
20,
21]. Several experimental studies have also shown that the administration of low-dose bioactive molecules is effective to obtain clean biological effects with a low probability of adverse effects.
To date, there are no therapies capable of blocking the neuronal death process that triggers neurodegenerative diseases. The use of the family of neurotrophic factors BDNF is part of, whose deficiency has been found in brain aging and in various pathological conditions, could become a valid therapeutic solution. However, there are still numerous problems to be solved, such as the calculation of the dose, the protocol of administration and the crossing of the BBB. Therefore, the aim of this study was to analyze the effect of low doses of BDNF on ROS production in both cultured astrocytes and cortical neurons by observing the behavior of endogenous antioxidant mechanisms. Furthermore, experiments were carried out in order to improve the tolerability of the substance by studying its ability to exert beneficial effects on the molecular pathways linked to viability in the nervous tissue. Finally, experiments were conducted both in vitro and in vivo to evaluate the characteristics of intestinal absorption after oral intake of BDNF and to evaluate the ability of BDNF to cross the BBB.
2. Materials and Methods
2.1. Preparation of BDNF Solutions
All dilutions were prepared starting from a stock solution (0.001 ng/mL) of BDNF 4CH in 0.9% NaCl. Based on previous knowledge on activated blends [
21,
22], BDNF solutions were prepared at two different concentrations: 1 pg/mL for in vitro studies and 1.20 pg/mL for in vivo studies. Each concentration was prepared using the sequential kinetic activation (SKA) method [
21]. These solutions are kinetically energized by a mechanically applied force via a standardized shaking process (sequential kinetic activation named SKA), characterized by vertical shaking corresponding to 100 oscillations in 10 s. All solutions were prepared by GUNA Laboratories (GUNA S.p.a, Milan, Italy). For each treatment, the volume of each solution was calculated by comparing the volume added to the sample treated with 50 ng/mL BDNF for in vitro study [
23] and with 25ng/mL BDNF [
24] for in vivo experiments. The BDNF used to compare the results obtained with BDNF 1 pg/mL SKA was not subjected to SKA treatment in order to replicate the same experimental conditions as in other studies.
2.2. Astrocytes Isolation
Primary mouse astrocyte cultures were extracted from C57BL/6 mouse pups, following a classical technique [
25] according to the National Guideline for the Use and Care of Laboratory Animals. Briefly, within 24 h of birth pups were euthanized, and cortices were dissected, minced, mechanically digested and left to settle for 30 min at room temperature. The cell suspension was centrifuged at 800 rpm for 5 min and pelleted cells were resuspended in Neuronal Basal Medium (Sigma-Aldrich, Milan, Italy) supplemented with 5% fetal bovine serum (FBS, Sigma-Aldrich, Milan, Italy), 1% penicillin/streptomycin (Sigma-Aldrich, Milan, Italy) and 2 mM L-glutamine (Sigma-Aldrich, Milan, Italy), plated in multi-wells and maintained in culture for 6 days before treatment. The cells were plated 4 × 104 astrocytes/cm
2 on a 24-well Transwell support to prepare the model BBB; 1 × 104 on 96-well plates for MTT and crystal violet staining; 4 × 104 cells were plated in black 96-well plates to study o × ygen consumption and mitochondrial membrane potential; 5 × 104 cells were plated on 24-well plates to analyze reactive o × ygen species (ROS) production; 2 × 105 cells were plated on 24-well plates to quantify BDNF; and 1 × 106 in 6-well plates to analyze the intracellular pathways by Western blot and ERK activity by ELISA test.
2.3. Primary Cortical Neuronal Cells
Primary mouse cortical neuronal cultures were obtained from the brains of P0 C57BL/6 mouse pups, as reported in literature [
26]. All procedures used in these studies follow the guidelines in accordance with the National Institutes of Health Guidelines. Cortices were dissected from embryonic brains and the tissue was mechanically dissociated and left to settle for 30 min at room temperature. After centrifuging only, the supernatant was re-suspended in Neuronal Basal medium (Sigma-Aldrich, Milan, Italy) supplemented with 2% B27 (Sigma-Aldrich, Milan, Italy), 1% penicillin/streptomycin (Sigma-Aldrich, Milan, Italy) and 2 mM L-glutamine (Sigma-Aldrich, Milan, Italy). Cells were plated on pre-coated plates with 10 µg/mL poly-L-lysine at a density of 1 × 106 cells/mL and were maintained in incubator at 37 °C with 5% CO2 and 95% humidity. At three days from plating, the medium was changed. All experiments were performed on primary cortical neuronal cells grown for 9–10 days in vitro. The cells were plated 1 × 104 on 96-well plates for MTT; 5 × 104 cells were plated on 24-well plates to analyze reactive oxygen species (ROS) production; 2 × 105 cells were plated on 24-well plates to quantify BDNF; and 4 × 104 cells were plated in black 96-well plates to study oxygen consumption and mitochondrial membrane potential.
2.4. In Vitro Experimental Protocol
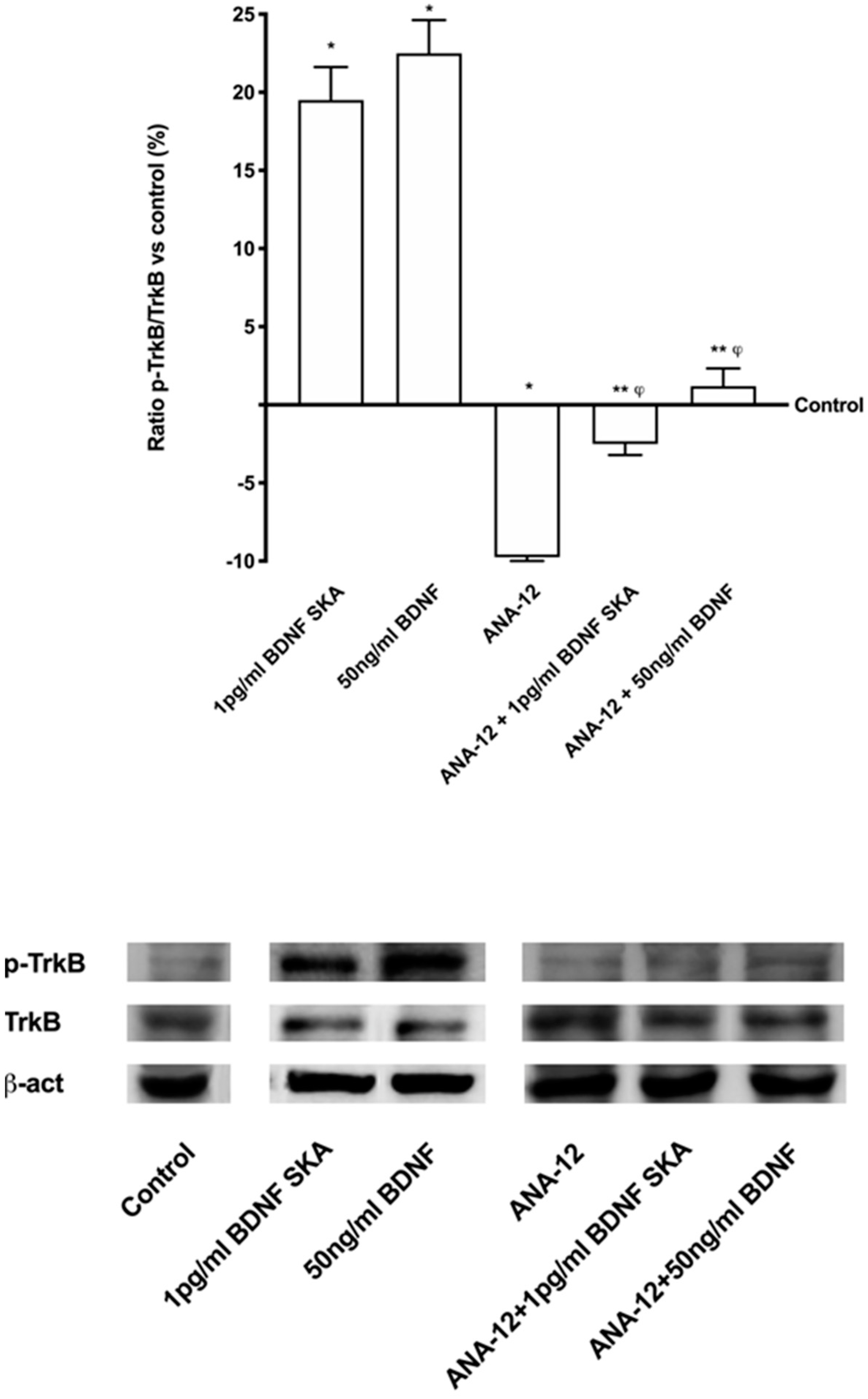
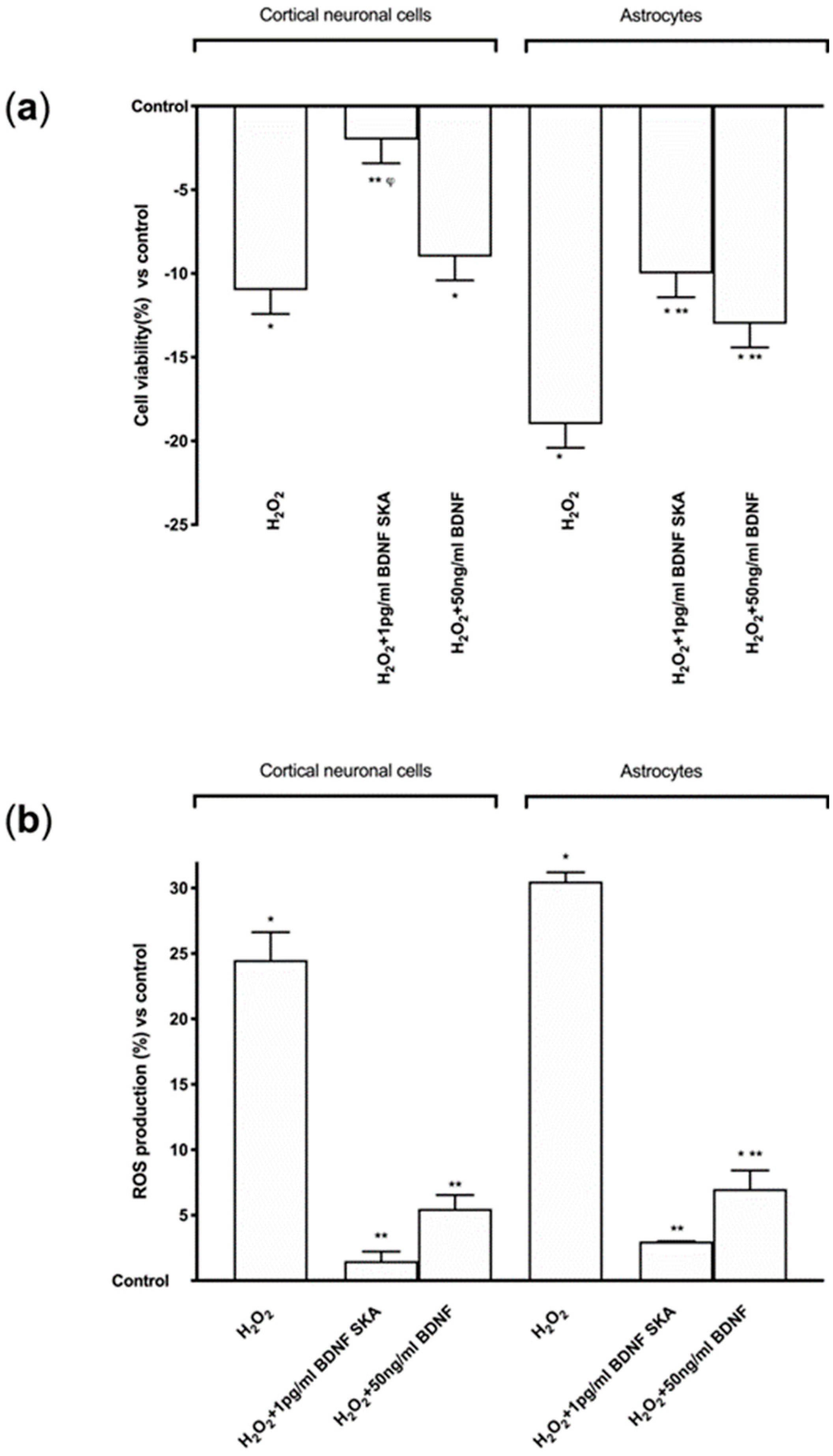
Before treatment, both primary cortical neuronal cells and astrocytes were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich, Milan, Italy) without red phenol and FBS, supplemented with 1% penicillin/streptomycin (Sigma-Aldrich, Milan, Italy) 2 mM L-glutamine (Sigma-Aldrich, Milan, Italy) and 1 mM sodium pyruvate (Sigma-Aldrich, Milan, Italy) at 37 °C, 5% CO2 and 95% humidity for 1 h. Cells were treated with 1 pg/mL BDNF SKA and 50 ng/mL BDNF at T0, checked every 24 h, and maintained for 6 days (named 6 days protocol). The vehicle, a saline solution, was also analyzed. Moreover, the involvement of TrkB using a specific antagonist, 1 µg/mL ANA-12 (Sigma-Aldrich, Milan, Italy) [
27] treating cells 30 min before stimulation was investigated. Additional experiments were carried out to analyze the ability of BDNF solutions to restore the damage caused by oxidative stress, a major cause of aging and neurodegeneration. Both cortical neuronal cells and astrocytes were pre-treated with 200 μM H
2O
2 (Sigma-Aldrich, Milan, Italy) [
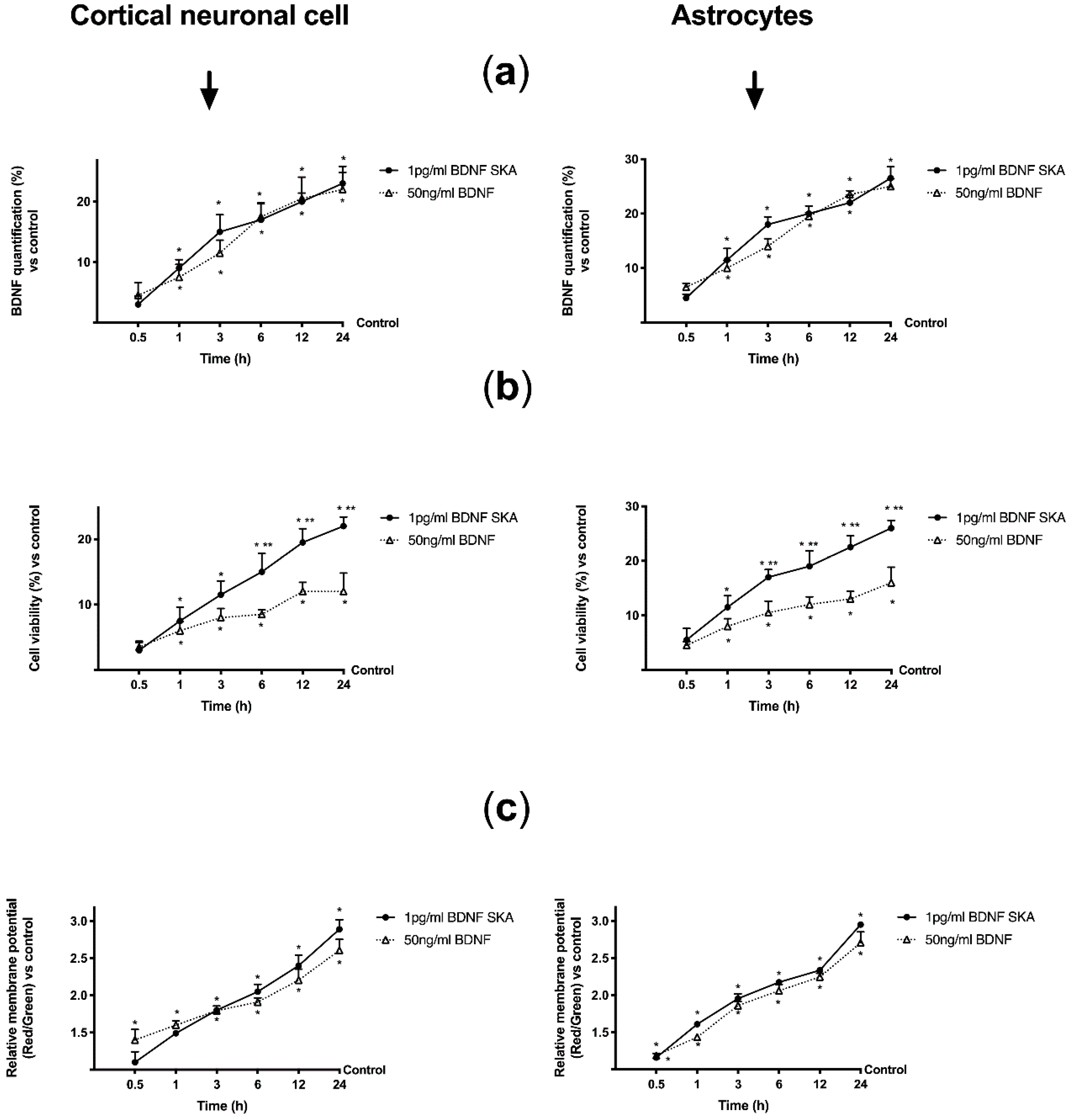
28] for 30 min and then treated with 1 pg/mL BDNF SKA and 50 ng/mL BDNF. Finally, astrocytes were used in time-course study within 24 h to mimic the human posology.
2.5. Intestinal Barrier In Vitro Model
Caco-2 cells (human epithelial colorectal adenocarcinoma cells), purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), were used as an experimental model [
29] to predict the features of intestinal absorption following oral intake [
30]. These cells were grown in a complete medium composed of Dulbecco’s Modified Eagle’s Medium/Nutrient F-12 Ham (DMEM-F12, Sigma-Aldrich, Milan, Italy) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich, Milan, Italy), 2 mM L-glutamine (Sigma-Aldrich, Milan, Italy), 1% penicillin-streptomycin (Sigma-Aldrich, Milan, Italy) and maintained in an incubator at 37 °C with 5% CO2 and 95% humidity. Cells were used from passages 46 to 49 and seeded in 24-well polyester Corning
® Costar
® transwell plates (Sigma-Aldrich, Milan, Italy) in complete medium. The cells were cultured for up to 21 days in a humidified incubator maintained at 37 °C in an atmosphere of 5% CO2, changing medium every 3 days first basolaterally and then apically. The monolayer integrity was checked every 3 days (at the time of the medium change) [
31]. After 21 days, 1pg/mL BDNF SKA and 50 ng/mL BDNF were added to culture medium under different pH conditions, as reported in literature [
31]; pH 6.5 preparations were added to the apical side, whereas pH 7.4 was added to the basolateral side. The slightly acidic pH (pH 6.5) in the apical side represents the average pH in the lumen of the small intestine, whereas the neutral pH (pH 7.4) in the basolateral side mimics the pH of the blood. During treatments, the cells were maintained in an incubator at 5% CO2, and at the end of stimulations the BDNF quantity was measured by ELISA kit after 30 min and 1, 3, 4, 5and 6h from stimulation. This model is suitable to predict the absorption of substances after oral intake by evaluating the apparent permeability coefficient (Papp) [
31,
32]. Briefly, the Papp (cm/s) was calculated as [
29]:
Papp = dQ/dt × 1/m0 × 1/A × VDonor
dQ: amount of substance transported (nmol or μg)
dt: incubation time (sec)
m0: amount of substrate applied to donor compartment (nmol or μg)
A: surface area of transwell membrane (cm2)
VDonor: volume of the donor compartment (cm3)
Negative controls without cells were tested to exclude transwell membranes influence.
2.6. Blood–Brain Barrier (BBB) Experimental Model
Astrocytes were co-cultured with human umbilical vein endothelial cells (HUVEC) cells according to methods reported in literature [
33]. HUVEC were purchased from ATCC
®. Cells were cultured in EGM Media (Lonza, Basel, Switzerland) supplemented with 10% FBS (Sigma-Aldrich, Milan, Italy), 1% penicillin/streptomycin (Sigma-Aldrich, Milan, Italy) and 2 mM Glutamine (Sigma-Aldrich, Milan, Italy) at 37 °C in a humidified atmosphere of 95% air, 5% CO2. In brief to create the BBB barrier, 4 × 104 astrocytes/cm
2 were plated on the basolateral side of the flipped 6.5 mm Transwells with polyester membrane with 0.4 μm pore size (Corning Costar, Sigma-Aldrich, Milan, Italy) and left to attach for 4 h. Transwells were then placed into the normal orientation and the cells left to grow for 48 h. After this time, 1 × 105 HUVEC cells/cm
2 were plated in the apical compartment. The inserts were then placed in a 24-well plate. After 7 days of culture, the Transwells were treated and permeability studies were performed [
34]. To understand the ability of tested substances to cross the blood–brain barrier the medium at the bottom side of the Transwells was quantified over time by measuring the volume and the concentration of BDNF.
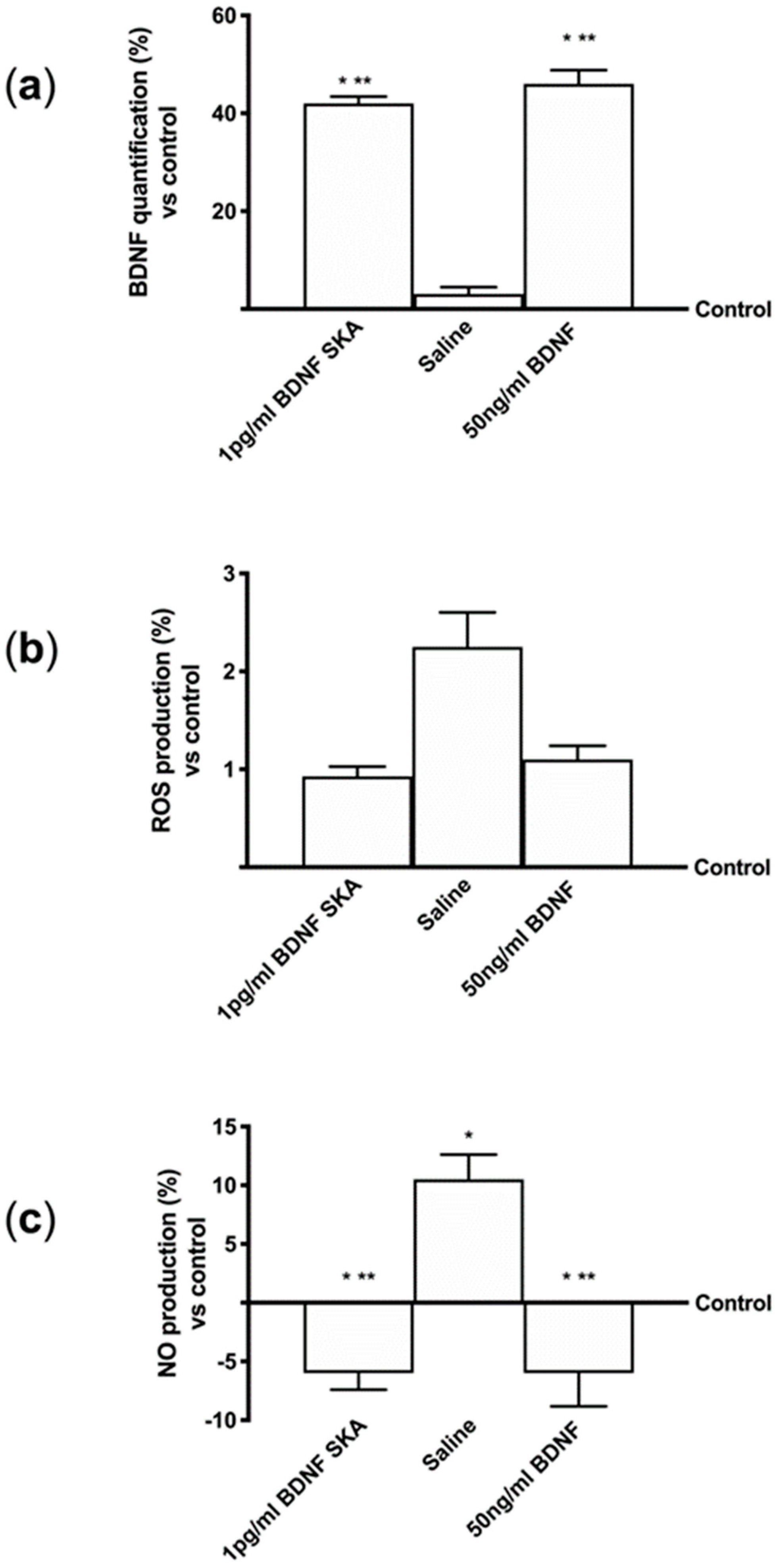
2.7. Brain-Derived Neurotrophic Factor (BDNF) Quantification
Brain-derived neurotrophic factor (BDNF) quantification was measured by Rat BDNF Elisa Kit (Thermo ScientificTM, Waltham, MA, United States) in cellular supernatants obtained from basolateral environment of BBB, primary cortical neuronal cells, astrocytes, serum and brain tissue to quantify BDNF, following the manufacturer’s instructions. Tissues were washed in saline solution, weighed, cut in small pieces and homogenized 100 mg tissue/300 μL with cold lysis buffer (0.1 M Tris, 0.01 M NaCl, 0.025 M EDTA, 1% NP40, 1% Triton X-100; Sigma-Aldrich, Milan, Italy) supplemented with 2 mM sodium orthovanadate, 0.1 M sodium fluoride (Sigma-Aldrich, Milan, Italy), 1:100 mix of protease inhibitors (Sigma-Aldrich, Milan, Italy), and 1:1000 phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich, Milan, Italy), using an electric potter at 1600 rpm for 2 min. The tissue extracts were centrifuged at 13000 rpm for 20 min at 4 °C.
The cellular and tissue supernatants were collected, and each sample was tested by ELISA kit. Briefly, biotinylated detection antibody was added into each well and the plate was incubated for 1 h at room temperature. Then, after 45 min of incubation with HRP-conjugated streptavidin, TMB substrate solution was added for 30 min and subsequently the reaction was stopped by adding Stop Solution. BDNF concentration was determined by measuring the absorbance through a spectrometer (VICTOR X4, multilabel plate reader) at 450 nm and calculated by comparing results to BDNF standard curve.
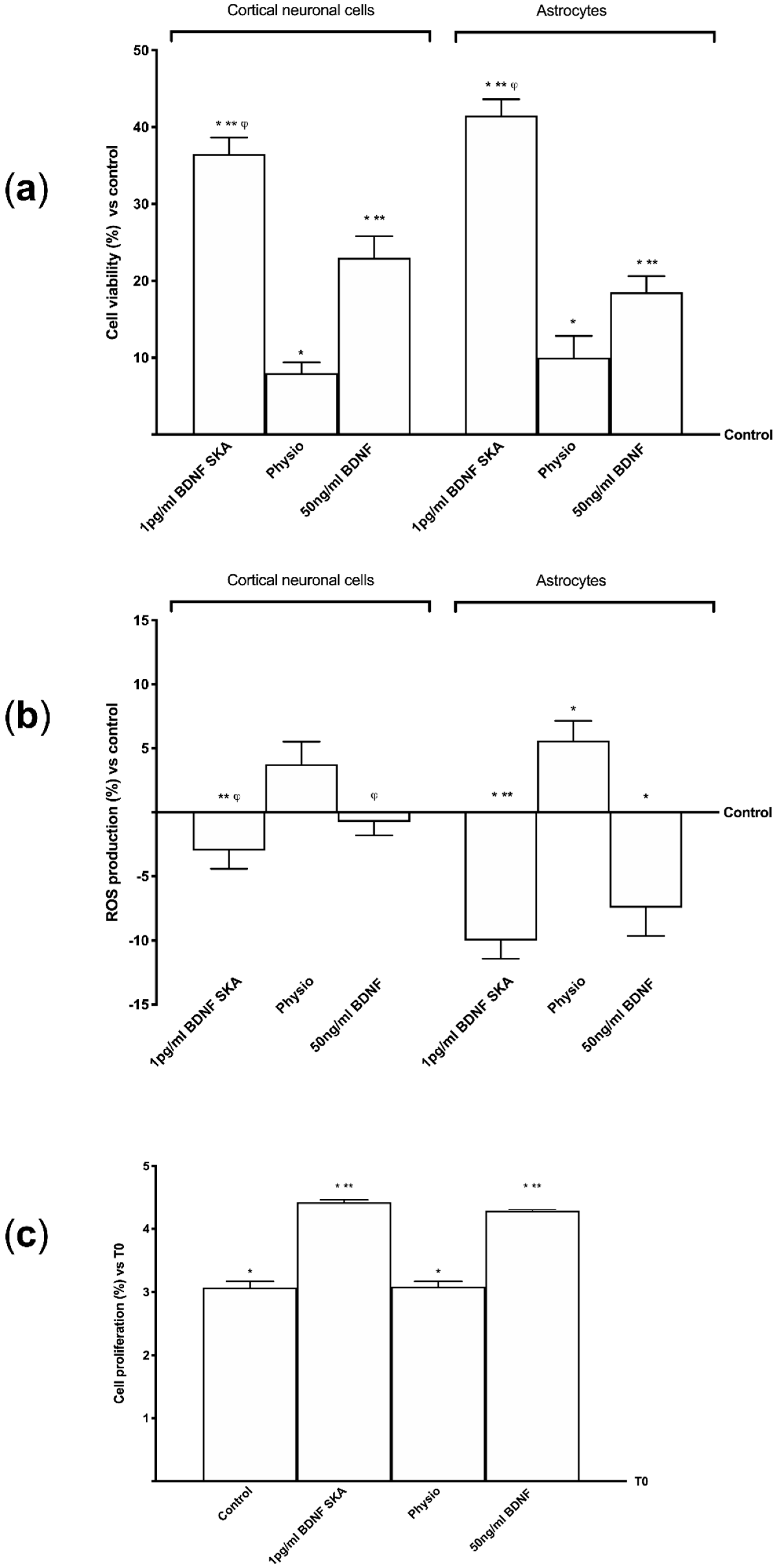
2.8. MTT Assay
MTT-based In Vitro Toxicology Assay Kit (Sigma-Aldrich, Milan, Italy) was performed on both cell types to determine cell viability, as previously described [
35]. Briefly, at the end of each stimulation, the cells were incubated with 1% MTT dye for 2 –3 h at 37 °C in incubator, until the purple crystals were dissolved in equal volume of MTT Solubilization Solution. The relative viability (%) was based on absorbance measuring through a spectrometer (VICTOR X4, Multilabel Plate Reader) at 570 nm with correction at 690 nm. Finally, viability was calculated comparing results to control cells (defined as 100% viable).
2.9. Crystal Violet Staining
After each treatment astrocytes were fixed with 1% glutaraldehyde (Sigma-Aldrich, Milan, Italy) for 15 min at room temperature, washed, and stained with 100 µL 0.1% aqueous crystal violet (Sigma-Aldrich, Milan, Italy) for 20 min at room temperature. To multi-well plates, 100 µL of 10% acetic acid was added and mixed before reading the absorbance at 595 nm using a spectrometer (VICTOR X4, multilabel plate reader). The estimated number was calculated by comparing the results to the control cells counted at T0.
2.10. ROS Production
The rate of reactive species of oxygen (ROS) was measured using a standard protocol based on the addition of cytochrome C (Sigma-Aldrich, Milan, Italy) to the samples and to another sample of 100 μL superoxide dismutase (Sigma-Aldrich, Milan, Italy). They were added for 30 min in an incubator at 37 °C, 5% CO
2, and 95% humidity to determine the antioxidant capability of BDNF solutions on the BBB model, primary cortical neuronal cells and astrocytes. At the end of stimulations, 100 µL of supernatant were measured at 550 nm using a spectrometer (VICTOR X4, multilabel plate reader) and O
2 was expressed as the mean ± SD (%) of nanomoles per reduced cytochrome C per micrograms of protein compared to control [
36].
2.11. NO Production
Nitric Oxide (NO) production was measured by Griess Assay (Promega Corporation, Madison, Wisconsin, United States). After 6 days of treatment, supernatants of basolateral BBB were mixed with equal volumes of Griess reagent and incubated in the dark at room temperature for 10 min; absorbance was measured by a spectrometer at 570 nm. NO production corresponded to the NO (μmol) produced after each stimulation by samples, each containing 1.5 μg of protein [
37].
2.12. Mitochondrial Membrane Potential
The mitochondrial membrane potential was analyzed following manufacturer’s instructions of Oxygen Consumption/Mito membrane Potential Dual Assay Kit (Cayman Chemical Company, Ann Arbor, MI, United States) [
38]. The mitochondrial membrane potential was measured using JC-1 aggregates at an excitation/emission of 560/590 nm and monomers at an excitation/emission of 485/535 nm in a fluorescence spectrometer (VICTOR X4, multilabel plate reader). The results are expressed as means ± SD (%) compared to control cells in both cell types.
2.13. ERK Activation Assay
ERK/MAPK activity was measured by the InstantOne™ ELISA (Thermo Fisher, Milan, Italy) on astrocytes lysates following the manufacturer’s instructions [
36]. Briefly, 50 μL/well of astrocytes lysed with cell lysis buffer were tested in InstantOne ELISA microplate strips after 1 h at room temperature on a microplate shaker with the antibody cocktail. At the end, the detection reagent was added for 20 min and then stopped by adding stop solution. The strips were measured by a spectrometer (VICTOR X4 multilabel plate reader) at 450 nm. The results were expressed as mean absorbance (%) compared to control.
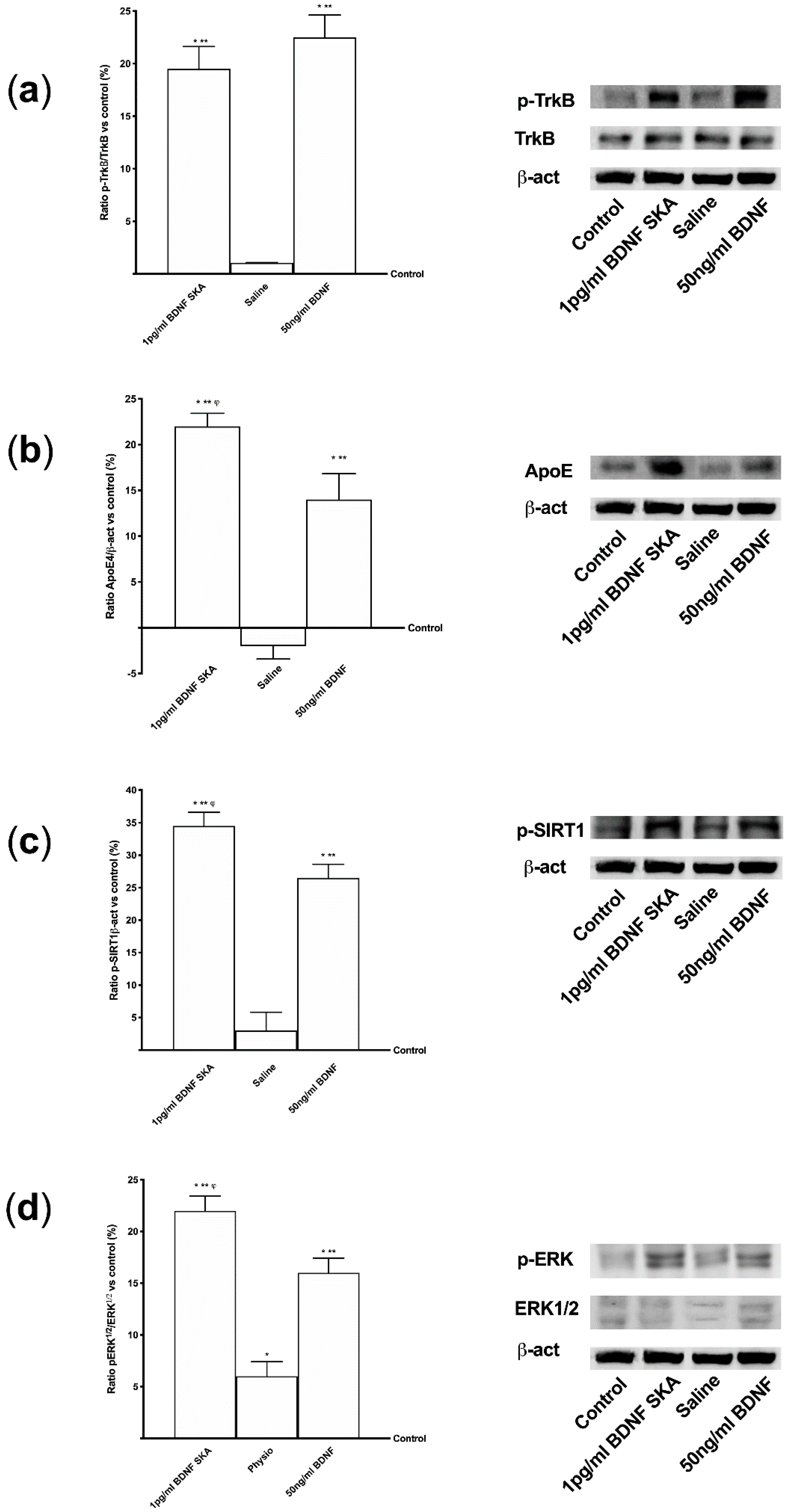
2.14. Western Blot
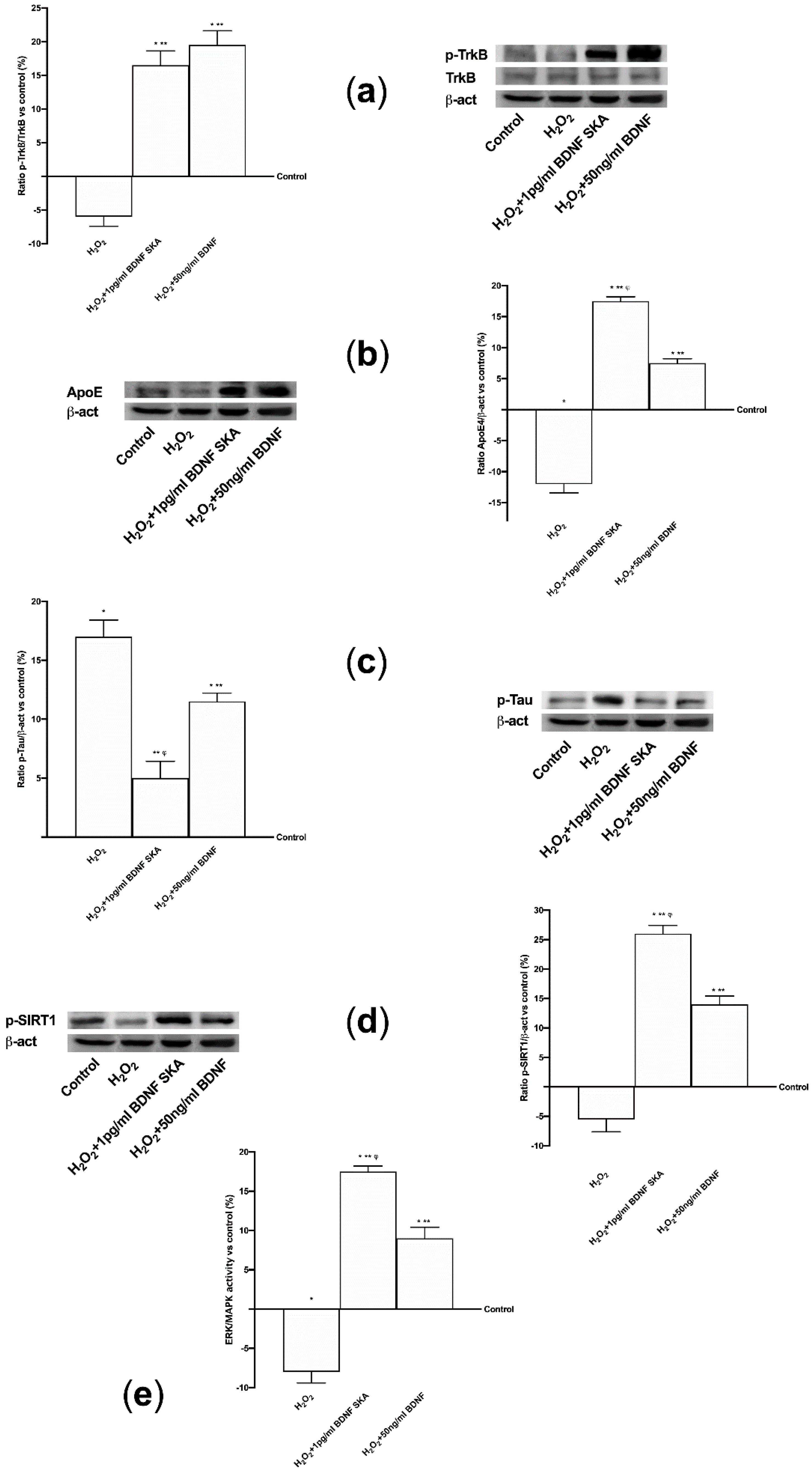
To perform Western blot analysis, 1 × 106 astrocytes plated in 6-well were lysed in ice with Ripa Buffer (50 mM Hepes, 150 mM NaCl, 0,1% SDS, 1% Triton X-100, 1% deoxycholate acid, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA, 1 mM NaF; Sigma-Aldrich, Milan, Italy) supplemented with 2 mM sodium orthovanadate and 1:100 mix Protease Inhibitor Cocktail (Sigma-Aldrich, Milan, Italy). 30 µg proteins were resolved on 8% and 15% SDS-PAGE gels. Brain tissue was also analyzed by Western blot to verify the mechanisms observed in cell culture. At the end of treatments brain tissue was excised out, washed in ice saline solution, weighed, cut in small pieces, and homogenized 100 mg tissue/300 µL with cold lysis buffer (0.1 M Tris, 0.01 M NaCl, 0.025 M EDTA, 1% NP40, 1% Triton X-100; Sigma-Aldrich, Milan, Italy) supplemented with 2 mM sodium orthovanadate, 0.1 M sodium fluoride (Sigma-Aldrich, Milan, Italy), 1:100 mix of protease inhibitors (Sigma-Aldrich, Milan, Italy), and 1:1000 phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich, Milan, Italy), using an electric potter at 1,600 rpm for 2 min. The tissue extracts were centrifuged at 13,000 rpm for 20 min at 4 °C and 40µg of each lysate were resolved on 8% and 15% SDS- PAGE gel. Polyvinylidene difluoride (PVDF) membranes (GE Healthcare Europe GmbH, Milan, Italy) obtained from cell and brain tissue lysates were incubated overnight at 4 °C with specific primary antibody: anti-phospho-tyrosine receptor kinase B (p-TrkB, Tyr515; 1:250, Santa Cruz, CA, United States), anti-tyrosine receptor kinase B (trkB; 1:250, Santa Cruz, CA, United States), anti-Apolipoprotein E (apoE, E4; 1:250, Santa Cruz, CA, United States), anti-phospho-Sirtuin1 (pSIRT1, Ser47; 1:1000, Sigma-Aldrich, Milan, Italy), anti-Phospho-p44/p42 Mitogen-activated protein kinase (pERK/MAPK, Thr202/Tyr204; 1:1000, Euroclone, Milan, Italy), anti-p44/p42 Mitogen-activated protein kinase (ERK/MAPK; 1:1000, Euroclone, Milan, Italy) and anti-Phospho-Tau (pTau, Ser262; 1:250, Thermo Fisher Scientific, Waltham, MA, United States). In addition, in brain anti-BDNF (1:500, Sigma-Aldrich, Milan, Italy) and anti-β-Amyloid (APP, B-4, 1:500, Santa Cruz, CA, United States) were also investigated. All protein expressions were normalized to the specific total protein (if possible), verified through β-actin detection (1:5000, Sigma-Aldrich, Milan, Italy) and expressed as mean ± SD (%).
2.15. Animal Model
12-month-old mice wild type C57BL/6jOlaHsd of comparable age to an elderly human (about 80 years old) [
39] purchased from Envigo++++ (Bresso, Italy), were used to confirm the effects of BDNF solutions in a complex model (
n = 52). Starting from a new protocol to induce a spontaneous intake [
40], we created a new rissole without bromophenol blue containing 1.2 pg/mL BDNF SKA or 25 ng/mL BDNF which is voluntary eaten by old mice. The quantity of rissoles was calculated considering the quantity of food and daily water normally taken by the animals [
41]. The rissole preparation phases are summed up in
Appendix A. The animals had access to food and water ad libitum and the experimental subjects were transferred to a single cage and kept in a single holding room and housed in a constant temperature of 21–22 °C, humidity of 5–55%, for 3 h [
40,
42]. Due to the short time taken to administer the rissole, mice showed no signs of social deprivation, such as increased aggressiveness. These signs have in fact been observed for periods of social deprivation of 6 h [
43]. After this time, the rissole was added in the lower part of the cage, but the mouse had free access to food and water in the upper part. Time of stimulation started from the addition of the rissole. During the whole period of treatment, the mice were monitored to assess their health status. Serum was obtained from blood of intracardiac withdrawal after inducing anesthesia, and brain tissue was obtained after animal death. All experimental procedures on animals were reviewed and approved by the University Committee OPBA (Organismo preposto al benessere degli animali) in accordance with local ethical standards and protocols approved by national guidelines (Approval No. 41/2019-PR). Animals were randomized into four different times of treatment: untreated (12 animals, 4 for each times of treatment), 24 h (20 animals), 24 h plus 24 h (10 animals) and 6-day protocol (only one administration for 6 days, 10 animals). In particular, 24 animals were sacrificed after 24 h and 14 animals were sacrificed after 48 h (24 h with rissole plus 24 h without rissole administration but the animals had access to food and water ad libitum in the upper part of cage). Finally, to demonstrate the efficacy of treatment, 14 animals were sacrificed 6 days after the only administration of BDNF solution (6-day protocol). All groups were sacrificed at specific time points (24 h, 24 h plus 24 h and 6 days) by CO
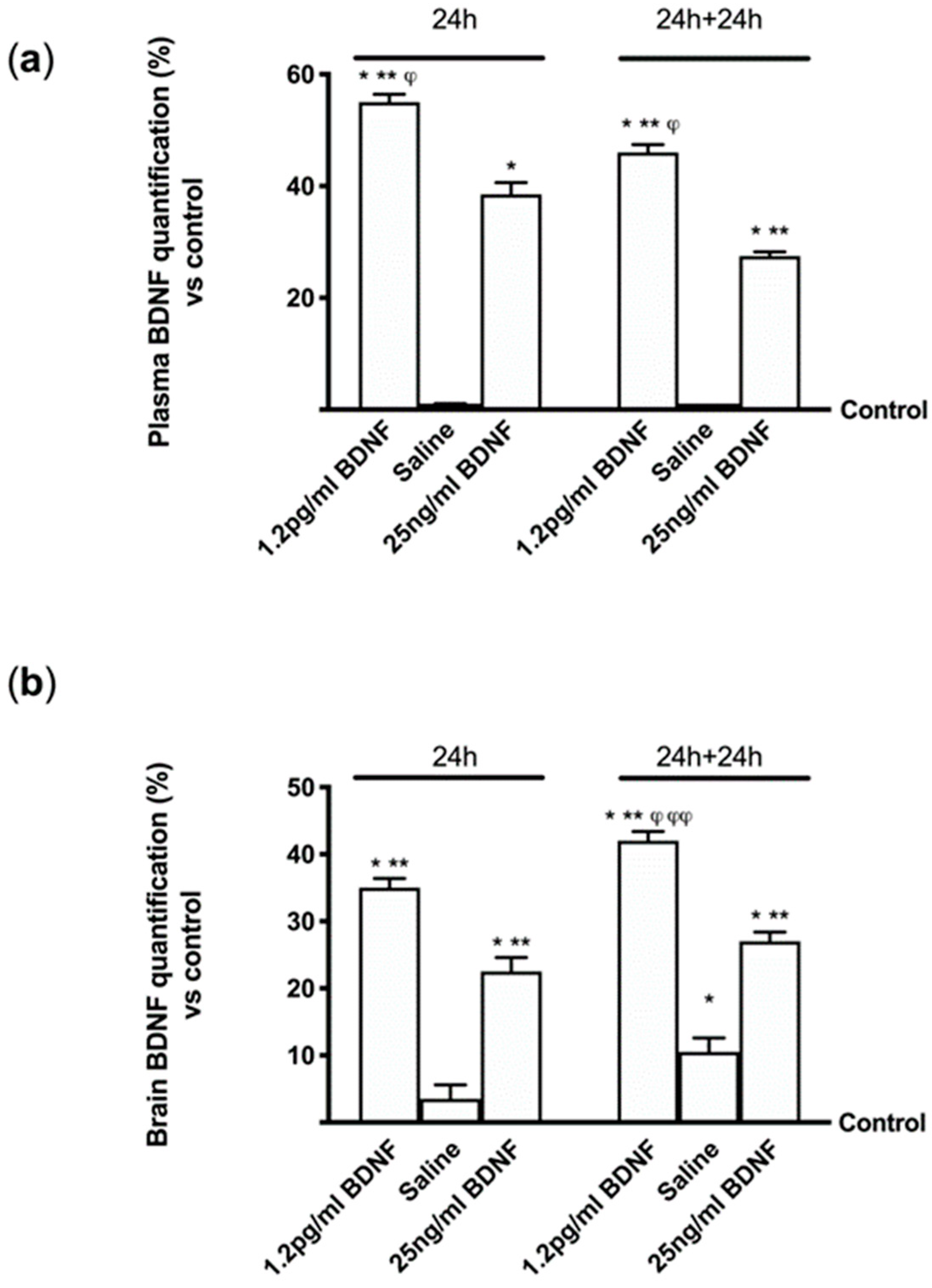
2 asphyxiation and blood drawn at the same time. The blood was centrifuged at 3500 rpm for 15 min at room temperature and the serum was conserved at −80 °C for subsequent experiments. In addition, the brain was removed, frozen and conserved at −80 °C for successive analysis by ELISA and Western blot on whole brain tissue lysates.
2.16. Statistical Analysis
Each part of the study is supported by at least 4 independent experiments both in vitro and in vivo. All results were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test; data are expressed as mean ± SD of at least four independent experiments for each experimental protocol produced in triplicates. The percentage values were compared through Mann–Whitney U test. Comparisons between the two groups were performed using a two-tailed Student’s t-test. Multiple comparisons between groups were analyzed by two-way ANOVA followed by a two-sided Dunnett post hoc testing. p-Value < 0.05 was considered statistically significant.
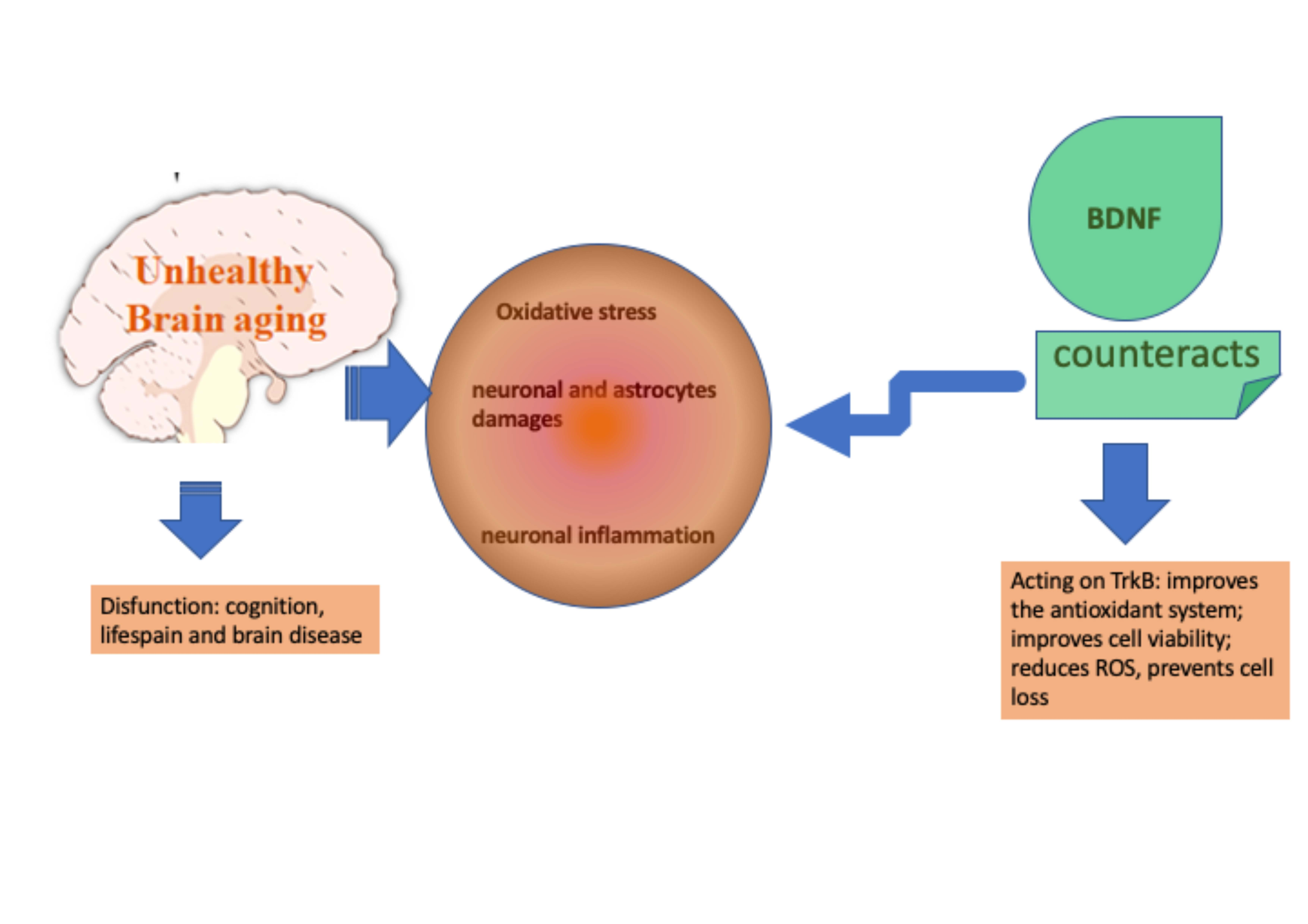
4. Discussion
Today neurodegenerative disorders are considered chronic and incurable conditions, whose disabling effects can last for years or decades. This represents a huge burden of suffering for patients and costs for health organizations. Optimal cognitive functions are linked to an efficient neuronal plasticity and the ability of neurons or glial cells to improve the efficacy of the synapses through biochemical and morphological changes, both at a dendritic and axonal level [
44]. However, as reported by many studies, this ability shows a marked age-related decrease [
15]. At present, treatments available for these diseases are mostly symptomatic or palliative and include neurotransmitter modulators, hormonal therapies, anti-inflammatory drugs, deep brain stimulation and herbal products. Therefore, there is an urgent need to develop new solutions able to restore the physiological functions of the brain tissue. Moreover, one of the main problems concerning the administration of active ingredients into the central nervous system is the crossing of the blood–brain barrier. Modern drug delivery systems can consist both of biodegradable and non-biodegradable formulations, which offer advantages in terms of protection, absorption, penetration and distribution of active ingredients. For this reason, the use of molecules already known for their exclusive functions within the brain and consequently physiologically predisposed to easily cross blood–brain barrier can be considered a valid option.
Recently, researchers’ attention has focused on the involvement of the neurotrophic factors in the development of neuronal decay. Currently, it is common knowledge that there are three main neurotrophic factors: the brain-derived neurotrophic factor (BDNF), the nerve growth factor and the glial cell-derived neurotrophic factor [
45]. BDNF in particular is associated with the modulation of neuroplasticity, which promotes the health of nervous tissue and also has the ability to counteract the effects of pro-inflammatory cytokines, which are key factors in neurodegenerative processes [
46].
In recent years, the possibility of using a growth factor therapy has been hypothesized by exploiting the growing information that research has accumulated, mainly on BDNF. Indeed, BDNF seems to have a real therapeutic potential, based on the observation that in many disorders of the nervous system serum BDNF levels are altered [
47]. However, a major problem is the delivery of the molecule to the affected cells. Although numerous studies have explored the possibility of administering BDNF through several approaches, such as gene therapy vectors, the development of mimetic peptides or even through direct administration into the nervous system, the results are still penalized by the lack of ease of use [
47]. Attempts to orally administer BDNF have so far yielded poor results due to the fact that BDNF is a moderately sized and charged protein and its transport through the intestinal barrier and BBB is not clear [
47].
The key idea of this work is to use a low dose BDNF solution to avoid the possible side effects of current therapies (such as sensitization and allergic reactions), supporting with experimental data a new potential therapeutic approach to treat or prevent neurodegenerative diseases. The concept of low-dose is an important and innovative aspect and has been shown to be effective in many studies. For example, in vivo treatment experiments with low-dose interferon were performed in many animal species [
48]. This treatment has been shown to induce dramatic clinical improvement in models of both infectious and chronic inflammatory diseases [
49].
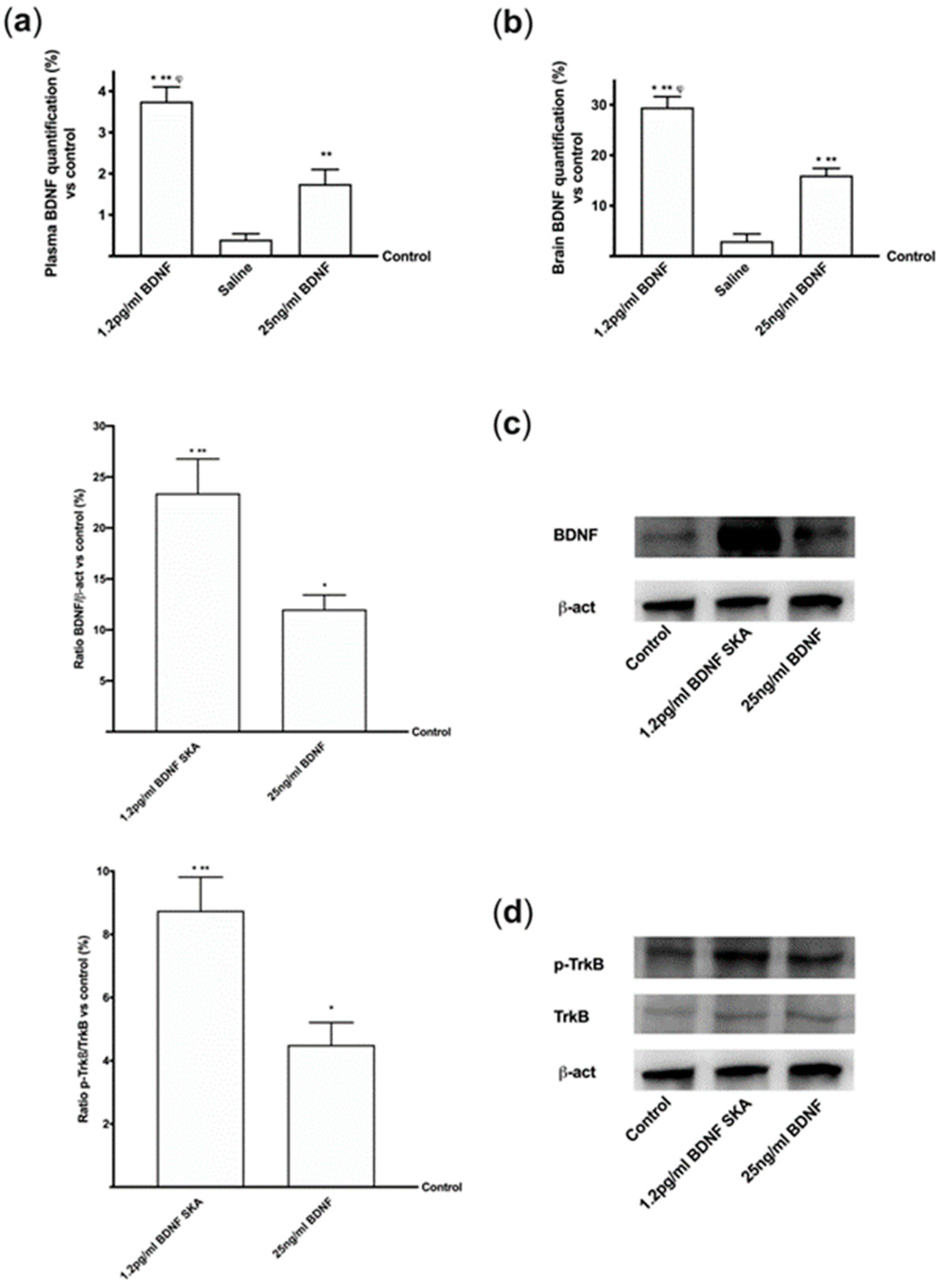
This work demonstrated, in in vitro experimental models, that BDNF is able to cross both the intestinal and BBB barrier, thus demonstrating the safety of its use.
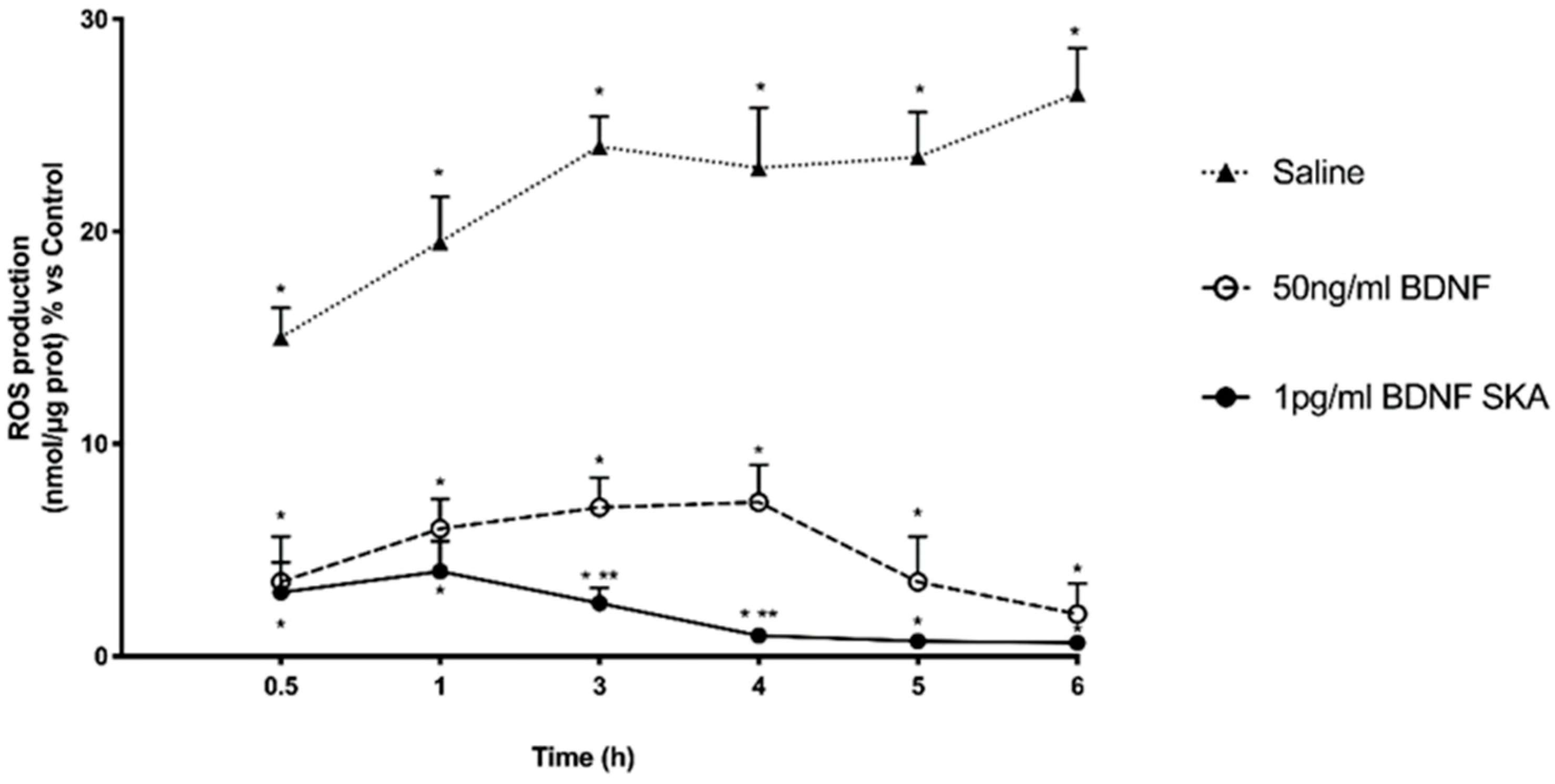
This study also demonstrates for the first time the efficacy of low-dose BDNF SKA in counteracting oxidative damage, which is one of the mechanisms underlying age-related neurodegeneration. The possibility of administering BDNF in very small amounts is a great advantage, both for the low risk of adverse effects and for the lower cost of treatment.
In the design of the study, it was decided to use a particular solution preparation technique, which is called SKA. It has been hypothesized that the mechanism of bioactive molecules, such as hormones, neuropeptides and growth factors, subjected to SKA and administered at low dosage, consists in the sensitization or activation of some cellular (or plasma) receptor units by virtue of their high dilution, and practically in their physiological working in the order of micrograms for hormones [
50] and picograms for the other messenger molecules [
51]. The SKA method has been used in previous works, which showed that SKA solutions have better biological effects than corresponding solutions that did not receive the same treatment [
20,
21,
29]. In the present study, it has been shown that BDNF SKA does not induce neuronal stress and it is able to counteract the formation of ROS.
During brain aging, the cells that show the first signs of degeneration are the astrocytes, despite the fact that subsequently the most important site of damage is represented by cortical neurons [
52]. BDNF SKA is able to increase cell viability in both neuronal cells and astrocytes, representing an important resource for the health of the nervous system.
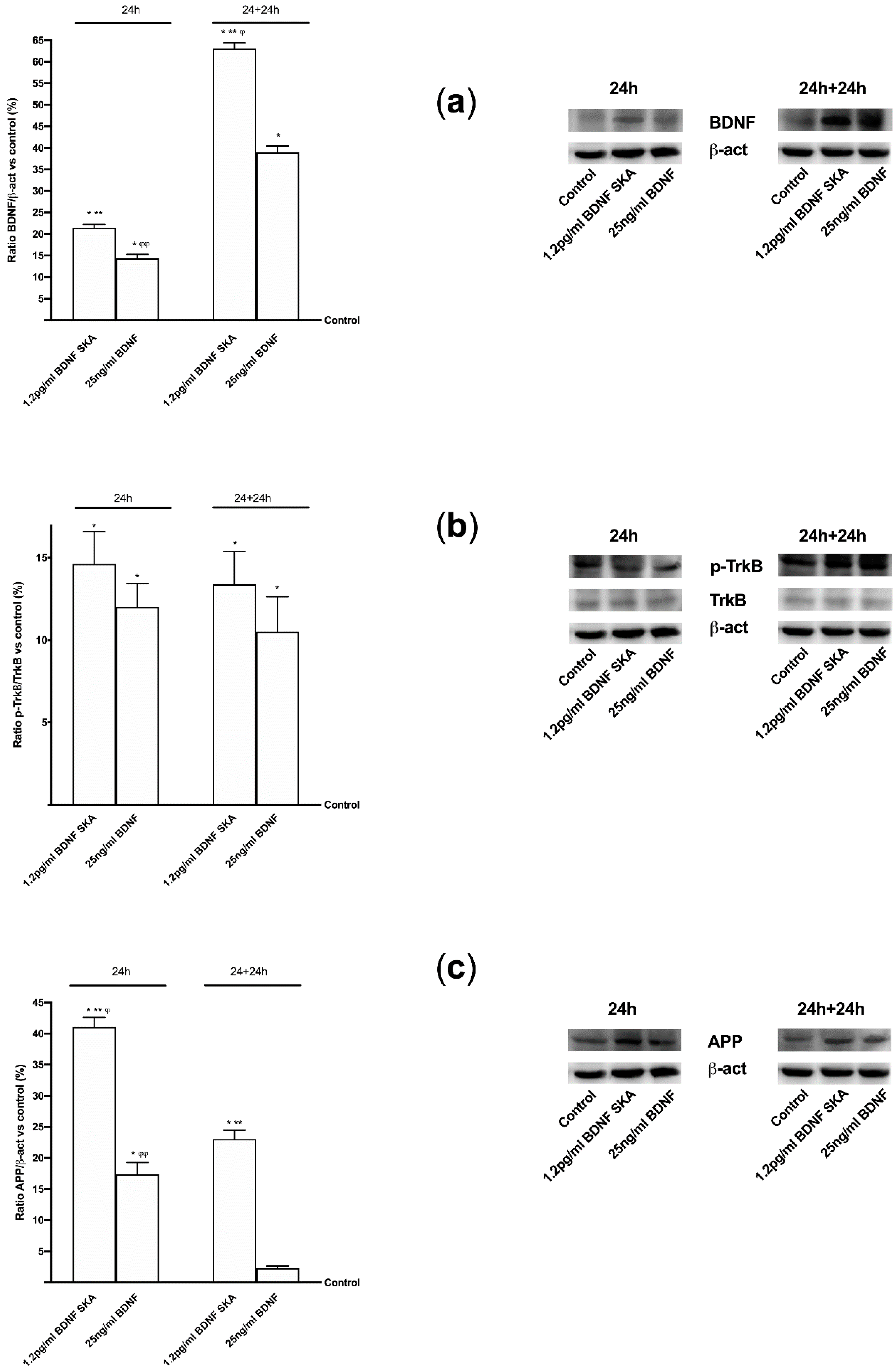
Furthermore, the aim of this study was to explore the timing of administration, to see if it was possible to identify an effective protocol that could be used in humans in the future. It can be hypothesized that the six-day protocol, consisting of a single administration followed by six days of measurements, stimulates cells without overloading normal physiological regulation. In this way a greater crossing of BDNF is achieved through the BBB, also inducing a low concentration of ROS and a reduced neuronal stress. In this way the normal cellular physiological processes are activated. The in vivo part of this research has allowed us to demonstrate that BDNF SKA has a high capacity to cross the enterohepatic circle and is able to remain in the bloodstream for at least 24 h. This makes the BDNF SKA a potential candidate for use as a food supplement. Since BDNF, necessary for the survival of neurons, is synthesized after the encoding of its specific gene, its expression was studied in this research. We observed that BDNF SKA at 24 h plus 24 h was able to induce the expression of endogenous BDNF, indicating a better effect of stimulation and induction of endogenous BDNF production. The beneficial effects of BDNF SKA have also been confirmed by the analysis of the amyloid protein precursor. Taken together, our results suggest that the simultaneous activation of at least ERK/MAPK is necessary to mediate a complete BDNF-dependent activation of the APP promoter [
53].
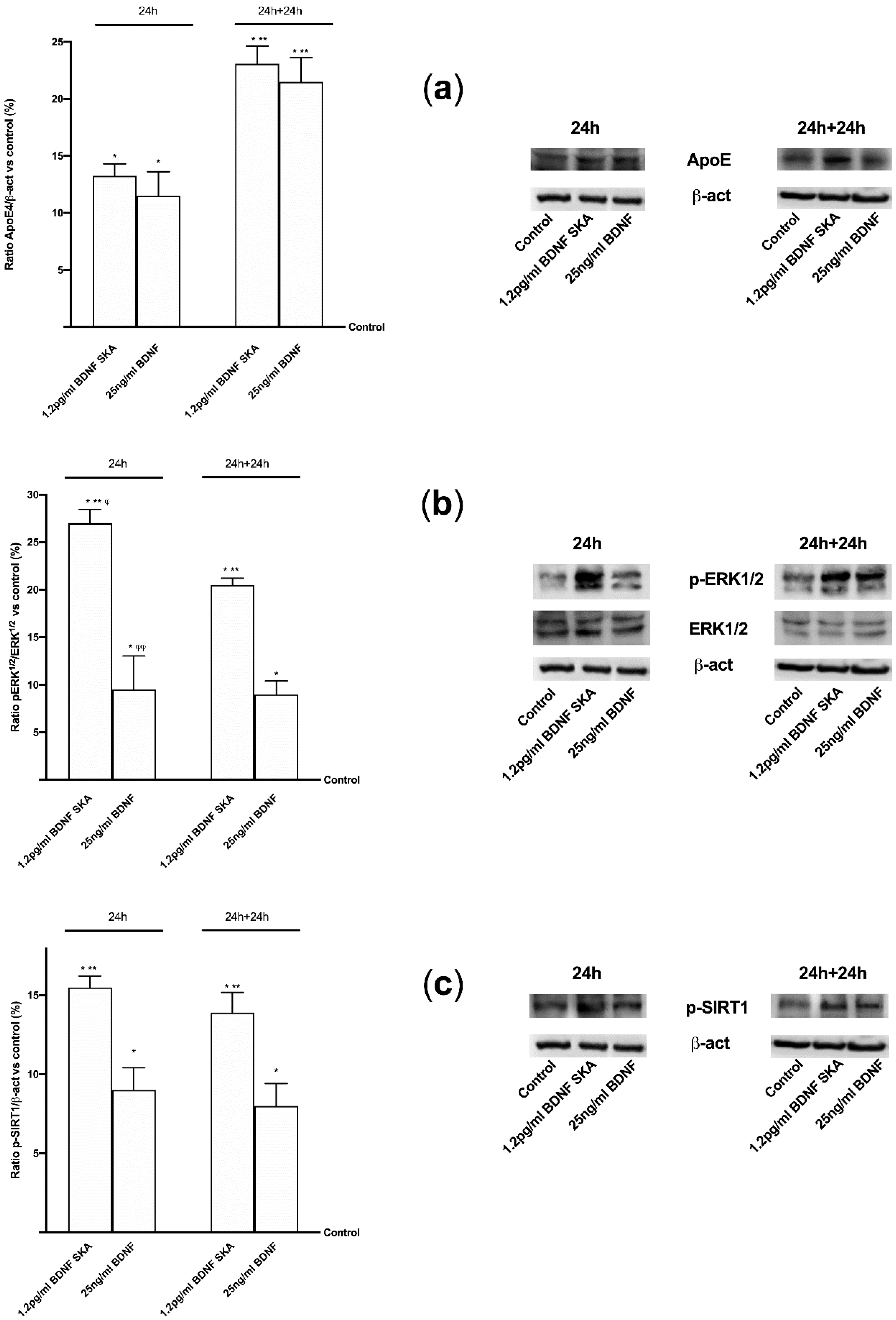
APP protein plays a central role in the development of Alzheimer’s disease; its expression, metabolism, splicing and secretion have been demonstrated to be regulated by ligands of the membrane tyrosine kinase receptors like BDNF [
54]. Furthermore, the study of the intracellular pathways demonstrated a significant increase in the expression of ApoE, which is a member of the low-density lipoprotein receptor gene family, mainly produced by the astrocytes in the brain. ApoE has been identified as the receptor that mediates amyloid β (Aβ) uptake and clearance by astrocytes, thus increasing glial LDLR levels, which may promote Aβ degradation within the brain [
55,
56,
57]; these data indicate a positive effect on brain trophism exerted by BDNF SKA and an increase in SIRT1 phosphorylation, confirming a potential role in counteracting the known mechanisms that lead to brain aging. Indeed, SIRT1 has recently been shown to play a role in normal cognitive function and synaptic plasticity, counteracting cognitive decline and neurodegenerative disease in aging [
58,
59]. The effectiveness of BDNF SKA has been carefully observed, allowing the serum BDNF levels to be maintained for up to six days after a single administration; these data suggest that BDNF SKA is able to remain in the nervous tissue for a long time even in the absence of treatment, triggering its own physiological production by cells better than high-dose BDNF. All our results support the hypothesis that treatment with BDNF SKA can protect the brain over time by inducing a physiological mechanism capable of slowing cell degeneration and may be a possible therapeutic strategy for the elderly population, in order to improve cognitive function. BDNF is also able to exert effects outside the nervous system, for example on the immune system [
60]. Therefore, interactions between the immune system and the effects described in this work could be hypothesized. The lack of this part could be a weak point in our research, which will be filled in subsequent research. On the other hand, one of the strengths of this study is the confirmation of the efficacy and safety of low-dose BDNF. The low-dose administration reduces the side effects of active molecules, without reducing their effectiveness is an important and innovative aspect. Indeed, the common clinical practice has long understood the essential importance of small stimuli compared to strong ones, which trigger self-regulatory and self-repairing mechanisms in the organism [
61]. In brain aging there is a decline in normal antioxidant defense mechanisms leading to increased brain vulnerability and finally to the deleterious effects of oxidative damage [
8]. Indeed, a large body of experimental research indicates that the brain is very susceptible to oxidative damage due to a high concentration of polyunsaturated fatty acids and transition metals that are involved in the generation of the hydroxyl radical [
62,
63]. Members of the ROS family, if not properly detoxified, start the process of oxidative damage, which can be defined as a chain reaction leading to sequential damage of all cellular components, in particular of lipids and proteins [
64]. Oxidative stress is considered one of the main mechanisms of cellular aging due to the ease of amplification of the damage and to the large number of target molecules [
8]. Cognitive deficits are the most common consequences of the aging process and they are characterized by massive neuronal loss, cognitive dysfunction and memory loss. Their incidence and prevalence continuously increase with advancing age [
65]. Indeed, it has been shown that low serum BDNF levels are linked to increased cognitive impairment [
66]. Moreover, BDNF helps to protect neurons from damage caused by infection or injury [
45] and participates in neuronal growth and maintenance and in different aspects of activity-dependent synaptic physiology by acting across different spatial and temporal domains [
67]. However, because of the difficulties associated with the administration of exogenous proteins into the central nervous system (CNS), it is important to consider the possibility of using endogenous sources of BDNF, for example, inducing increased glial cell activity. It is well known that glial cells increase expression of a variety of growth factors, including BDNF. In particular, in the adult brain, astrocytes are the cells responsible for maintaining neuronal and synaptic function [
68].


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}