Optimized Protocol to Generate Spinal Motor Neuron Cells from Induced Pluripotent Stem Cells from Charcot Marie Tooth Patients

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
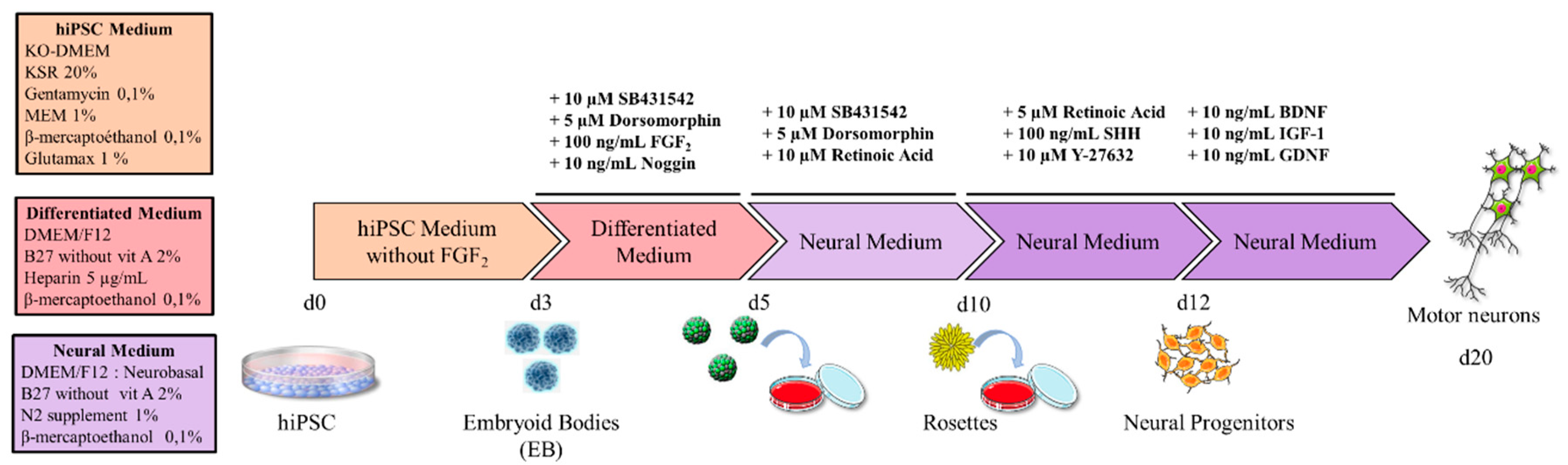
2.1. Cell Culture Media
2.2. Experimental Design
2.2.1. Generation of hiPSCs
2.2.2. Generation of Motor Neurons
2.3. Immunostaining
2.4. Electrophysiology
3. Results
3.1. Obtaining iPSCs from Patients
3.2. Definition of the Factors and the Timeframes for MN Differentiation
3.3. Differentiation into Motor Neurons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Statements
Appendix A. HiPSC Characterization
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-J.; Du, Z.-W.; Zarnowska, E.D.; Pankratz, M.; Hansen, L.O.; Pearce, R.A.; Zhang, S.-C. Specification of motoneurons from human embryonic stem cells. Nat. Biotechnol. 2005, 23, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Singh Roy, N.; Nakano, T.; Xuing, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Enhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cells. Exp. Neurol. 2005, 196, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [Green Version]
- Ebert, A.D.; Yu, J.; Rose, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Hu, B.-Y.; Zhang, S.-C. Differentiation of spinal motor neurons from pluripotent human stem cells. Nat. Protoc. 2009, 4, 1295–1304. [Google Scholar] [CrossRef]
- Kim, D.-S.; Lee, J.S.; Leem, J.W.; Huh, Y.J.; Kim, J.Y.; Kim, H.-S.; Park, I.-H.; Daley, G.Q.; Hwang, D.-Y.; Kim, D.-W. Robust Enhancement of Neural Differentiation from Human ES and iPS Cells Regardless of their Innate Difference in Differentiation Propensity. Stem Cell Rev. Rep. 2010, 6, 270–281. [Google Scholar] [CrossRef]
- Saporta, M.A.; Dang, V.; Volfson, D.; Zou, B.; Xie, X.S.; Adebola, A.; Liem, R.K.; Shy, M.; Dimos, J.T. Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 2015, 263, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciel, R.; Correa, R.; Bosso Taniguchi, J.; Prufer Araujo, I.; Saporta, M.A. Human Tridimensional Neuronal Cultures for Phenotypic Drug Screening in Inherited Peripheral Neuropathies. Clin. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Besser, R.R.; Bowles, A.C.; Alassaf, A.; Carbonero, D.; Claure, I.; Jones, E.; Reda, J.; Wubker, L.; Batchelor, W.; Ziebarth, N.; et al. Enzymatically crosslinked gelatin-laminin hydrogels for applications in neuromuscular tissue engineering. Biomater. Sci. 2020, 8, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wichterle, H.; Lieberam, I.; Porter, J.A.; Jessell, T.M. Directed Differentiation of Embryonic Stem Cells into Motor Neurons. Cell 2002, 110, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Miles, G.B.; Yohn, D.C.; Wichterle, H.; Jessell, T.M.; Rafuse, V.F.; Brownstone, R.M. Functional properties of motoneurons derived from mouse embryonic stem cells. J. Neurosci. 2004, 24, 7848–7858. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Cerrato, V.; Fucà, E.; Parmigiani, E.; Buffo, A.; Leto, K. Sonic hedgehog patterning during cerebellar development. Cell. Mol. Life Sci. 2016, 73, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Iams, W.T.; Lovly, C.M. Molecular pathways: Clinical applications and future direction of insulin-like growth factor-1 receptor pathway blockade. Clin. Cancer Res. 2015, 21, 4270–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Tassigny, X.d.; Pascual, A.; Lopez-Barneo, J. GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for parkinson’s disease. Front. Neuroanat. 2015, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Ueno, M.; Kamiya, D.; Nishiyama, A.; Matsumura, M.; Wataya, T.; Takahashi, J.B.; Nishikawa, S.; Nishikawa, S.; Muguruma, K.; et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007, 25, 681–686. [Google Scholar] [CrossRef]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maden, M. Retinoid signalling in the development of the central nervous system. Nat. Rev. Neurosci. 2002, 3, 843–853. [Google Scholar] [CrossRef]
- Powers, R.K.; Binder, M.D. Persistent sodium and calcium currents in rat hypoglossal motoneurons. J. Neurophysiol. 2003, 89, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Harfe, B.D.; Scherz, P.J.; Nissim, S.; Tian, H.; McMahon, A.P.; Tabin, C.J. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 2004, 118, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, S.; Sandler, O.; Sberro, H.; Shnider, S.; Schejter, E.; Shilo, B.-Z.; Barkai, N. Pre-steady-state decoding of the Bicoid morphogen gradient. PLoS Biol. 2007, 5, e46. [Google Scholar] [CrossRef] [Green Version]
- Gregor, T.; Tank, D.W.; Wieschaus, E.F.; Bialek, W. Probing the limits to positional information. Cell 2007, 130, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Andrews, M.G.; Kong, J.; Novitch, B.G.; Butler, S.J. New Perspectives on the Mechanisms Establishing the Dorsal-Ventral Axis of the Spinal Cord, 1st ed.; Elsevier Inc.: New York, NY, USA, 2019; Volume 132, ISBN 9780128104897. [Google Scholar]
- Ducy, P.; Karsenty, G. The family of bone morphogenetic proteins. Kidney Int. 2000, 57, 2207–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bier, E.; De Robertis, E.M. BMP gradients: A paradigm for morphogen-mediated developmental patterning. Science 2015, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.F. Developmental Biology, 6th ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 2000; p. 695. [Google Scholar]
- Casarosa, S.; Zasso, J.; Conti, L. Systems for Ex-Vivo Isolation and Culturing of Neural Stem Cells. Neural Stem Cells New Perspect. 2013. [Google Scholar] [CrossRef] [Green Version]
- Kudoh, T.; Wilson, S.W.; Dawid, I.B. Distinct roles for Fgf, Wnt and retinoic acid in posteriorizing the neural ectoderm. Development 2002, 129, 4335–4346. [Google Scholar] [PubMed]
- Davis-Dusenbery, B.N.; Williams, L.A.; Klim, J.R.; Eggan, K. How to make spinal motor neurons. Development 2014, 141, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Cardenas-Aguayo, M.D.C.; Kazim, S.F.; Grundke-Iqbal, I.; Iqbal, K. Neurogenic and Neurotrophic Effects of BDNF Peptides in Mouse Hippocampal Primary Neuronal Cell Cultures. PLoS ONE 2013, 8, e53596. [Google Scholar] [CrossRef]
- Jung, H.J.; Suh, Y. Regulation of IGF −1 signaling by microRNAs. Front. Genet. 2015, 5, 472. [Google Scholar] [CrossRef] [Green Version]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef]
- Wrigley, S.; Arafa, D.; Tropea, D. Insulin-like growth factor 1: At the crossroads of brain development and aging. Front. Cell. Neurosci. 2017, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 2001, 12, 361–373. [Google Scholar] [CrossRef]
- Timmerman, V.; Strickland, A.V.; Züchner, S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes Basel 2014, 5, 13–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juneja, M.; Burns, J.; Saporta, M.A.; Timmerman, V. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry 2019, 90, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Juneja, M.; Azmi, A.; Baets, J.; Roos, A.; Jennings, M.J.; Saveri, P.; Pisciotta, C.; Bernard-Marissal, N.; Schneider, B.L.; Verfaillie, C.; et al. PFN2 and GAMT as common molecular determinants of axonal Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Ohara, R.; Imamura, K.; Morii, F.; Egawa, N.; Tsukita, K.; Enami, T.; Shibukawa, R.; Mizuno, T.; Nakagawa, M.; Inoue, H. Modeling Drug-Induced Neuropathy Using Human iPSCs for Predictive Toxicology. Clin. Pharmacol. Ther. 2017, 101, 754–762. [Google Scholar] [CrossRef]
- Kim, J.Y.; Woo, S.Y.; Hong, Y.B.; Choi, H.; Kim, J.; Choi, H.; Mook-Jung, I.; Ha, N.; Kyung, J.; Koo, S.K.; et al. HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with HSPB1 Mutation. Stem Cells Int. 2016, 2016, 9475981. [Google Scholar] [CrossRef] [Green Version]
- Faye, P.A.; Poumeaud, F.; Miressi, F.; Lia, A.S.; Demiot, C.; Magy, L.; Favreau, F.; Sturtz, F.G. Focus on 1,25-dihydroxyvitamin D3 in the peripheral nervous system. Front. Neurosci. 2019, 13, 348. [Google Scholar] [CrossRef]
- Faye, P.-A.; Vedrenne, N.; De la Cruz-Morcillo, M.A.; Barrot, C.-C.; Richard, L.; Bourthoumieu, S.; Sturtz, F.; Funalot, B.; Lia, A.-S.; Battu, S. New Method for Sorting Endothelial and Neural Progenitors from Human Induced Pluripotent Stem Cells by Sedimentation Field Flow Fractionation. Anal. Chem. 2016, 88, 6696–6702. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Cat Num | Species/Type | Dilution |
|---|---|---|---|---|
| Pluripotency | ||||
| Nanog | Abcam | 130095632 | Rabbit poly IgG | 1:100 |
| Oct3/4 | Santa Cruz Biotech | sc-5279 | Mouse Mono IgG2B | 1:100 |
| Sox2 | Chemicon | AB5603 | Rabbit poly IgG | 1:100 |
| Spontaneous Differentiation in Three Germinal Layers | ||||
| Pax6 | Covance | PRB-278P | Rabbit poly IgG | 1:100 |
| αSMA | DAKO | M0851 | Mouse IgG2A | 1:500 |
| Sox17 | R&D | AF1924 | Goat IgG | 1:100 |
| Neuronal and Motor Neuronal | ||||
| Tuj1 | R&D | MAB1195 | Mouse Mono IgG | 1:500 |
| PGP9.5 | Ultraclone | Ra95101 | Rabbit poly IgG | 1:500 |
| HB = MNR2 | DSHB | 81.5C10 | Chicken | 1:100 |
| Islet1 | DSHB | 40.2D6-c | Mouse Mono IgG | 1:25 |
| Islet1/2 | DSHB | 39.4D5-c | Mouse Mono IgG | 1:25 |
| ChAT | Chemicon | AB144P | Goat IgG | 1:20 |
| Other | ||||
| Ki-67 | Leica | NCL-L-Ki67-MM1 | Mouse Mono IgG | 1:200 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faye, P.-A.; Vedrenne, N.; Miressi, F.; Rassat, M.; Romanenko, S.; Richard, L.; Bourthoumieu, S.; Funalot, B.; Sturtz, F.; Favreau, F.; et al. Optimized Protocol to Generate Spinal Motor Neuron Cells from Induced Pluripotent Stem Cells from Charcot Marie Tooth Patients. Brain Sci. 2020, 10, 407. https://doi.org/10.3390/brainsci10070407
Faye P-A, Vedrenne N, Miressi F, Rassat M, Romanenko S, Richard L, Bourthoumieu S, Funalot B, Sturtz F, Favreau F, et al. Optimized Protocol to Generate Spinal Motor Neuron Cells from Induced Pluripotent Stem Cells from Charcot Marie Tooth Patients. Brain Sciences. 2020; 10(7):407. https://doi.org/10.3390/brainsci10070407
Chicago/Turabian StyleFaye, Pierre-Antoine, Nicolas Vedrenne, Federica Miressi, Marion Rassat, Sergii Romanenko, Laurence Richard, Sylvie Bourthoumieu, Benoît Funalot, Franck Sturtz, Frederic Favreau, and et al. 2020. "Optimized Protocol to Generate Spinal Motor Neuron Cells from Induced Pluripotent Stem Cells from Charcot Marie Tooth Patients" Brain Sciences 10, no. 7: 407. https://doi.org/10.3390/brainsci10070407