
Systematic Review of Nicotine Exposure’s Effects on Neural Stem and Progenitor Cells



Abstract
:1. Introduction
2. Nicotine-Related Decreases in NSC Proliferation
3. Nicotine-Related Increases in NSC Proliferation
4. Nicotine Attenuates Aß-Induced Neurotoxicity in NSCs
5. Nicotine Induces Mitochondrial Stress in Neural Stem Cells
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Altman, J.; Das, G.D. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 1965, 124, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Reynolds, B.A.; Vescovi, A.L.; Morshead, C.; Craig, C.G.; Van Der Kooy, D. Is there a neural stem cell in the mammalian forebrain? Trends Neurosci. 1996, 19, 387–393. [Google Scholar] [CrossRef]
- Levison, S.W.; Goldman, J.E. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron 1993, 10, 201–212. [Google Scholar] [CrossRef]
- Kempermann, G.; Kuhn, H.G.; Gage, F.H. More hippocampal neurons in adult mice living in an enriched environment. Nature 1997, 386, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Beylin, A.; Tanapat, P.; Reeves, A.; Shors, T.J. Learning enhances adult neurogenesis in the hippocampal formation. Nat. Neurosci. 1999, 2, 260–265. [Google Scholar] [CrossRef]
- Shors, T.J.; Miesegaes, G.; Beylin, A.; Zhao, M.; Rydel, T.; Gould, E. Neurogenesis in the adult is involved in the formation of trace memories. Nature 2001, 410, 372–376. [Google Scholar] [CrossRef]
- Doetsch, F.; Caille, I.; Lim, D.A.; García-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Altman, J. Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J. Comp. Neurol. 1969, 137, 433–457. [Google Scholar] [CrossRef]
- Lois, C.; Alvarez-Buylla, A. Long-distance neuronal migration in the adult mammalian brain. Science 1994, 264, 1145–1148. [Google Scholar] [CrossRef]
- Lois, C.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Chain migration of neuronal precursors. Science 1996, 271, 978–981. [Google Scholar] [CrossRef]
- Lois, C.; Alvarez-Buylla, A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl. Acad. Sci. USA 1993, 90, 2074–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.A.; Alvarez-Buylla, A. The adult ventricular-subventricular zone (V-SVZ) and olfactory bulb (OB) neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8, a018820. [Google Scholar] [CrossRef] [PubMed]
- Valentine, G.; Sofuoglu, M. Cognitive Effects of Nicotine: Recent Progress. Curr. Neuropharmacol. 2018, 16, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Sutton, S.K.; Van Rensburg, K.J.; Jentink, K.G.; Drobes, D.J.; Evans, D.E. Nicotine-induced cortical activation among nonsmokers with moderation by trait cognitive control. Psychopharmacology 2016, 233, 2301–2308. [Google Scholar] [CrossRef]
- Warburton, D.M.; Rusted, J.M. Cholinergic control of cognitive resources. Neuropsychobiology 1993, 28, 43–46. [Google Scholar] [CrossRef]
- Hahn, B.; Stolerman, I.P. Nicotine-induced attentional enhancement in rats: Effects of chronic exposure to nicotine. Neuropsychopharmacology 2002, 27, 712–722. [Google Scholar] [CrossRef] [Green Version]
- Gandelman, J.A.; Kang, H.; Antal, A.; Albert, K.; Boyd, B.D.; Conley, A.C.; Newhouse, P.; Taylor, W.D. Transdermal Nicotine for the Treatment of Mood and Cognitive Symptoms in Nonsmokers With Late-Life Depression. J. Clin. Psychiatry 2018, 79, 79. [Google Scholar] [CrossRef]
- Alkadhi, K.A. Neuroprotective Effects of Nicotine on Hippocampal Long-Term Potentiation in Brain Disorders. J. Pharmacol. Exp. Ther. 2018, 366, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.; Li, R.; Lyketsos, C.; Livingston, G. Treatment for mild cognitive impairment: Systematic review. Br. J. Psychiatry 2013, 203, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Quik, M.; Perez, X.A.; Bordia, T. Nicotine as a potential neuroprotective agent for Parkinson’s disease. Mov. Disord. 2012, 27, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Adam, B.L.; Terry, A.V., Jr. Evaluation of nicotine and cotinine analogs as potential neuroprotective agents for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2014, 24, 1472–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majdi, A.; Kamari, F.; Vafaee, M.S. Sadigh-Eteghad S Revisiting nicotine’s role in the ageing brain and cognitive impairment. Rev. Neurosci. 2017, 28, 767–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Zheng, Q.; Liu, H.; Wan, T.; Zhou, J.; Li, D.; Zhou, H.; Li, J.; Ji, F.; Tang, W.; et al. Nicotine-Induced Neuroprotection against Cognitive Dysfunction after Partial Hepatectomy Involves Activation of BDNF/TrkB Signaling Pathway and Inhibition of NF-kappaB Signaling Pathway in Aged Rats. Nicotine Tob. Res. 2018, 20, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Takarada, T.; Nakamichi, N.; Kawagoe, H.; Ogura, M.; Fukumori, R.; Nakazato, R.; Fujikawa, K.; Kou, M.; Yoneda, Y. Possible neuroprotective property of nicotinic acetylcholine receptors in association with predominant upregulation of glial cell line-derived neurotrophic factor in astrocytes. J. Neurosci. Res. 2012, 90, 2074–2085. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.R.; Van Der Schyf, C.J.; Abbruscato, T.J.; Allen, D.D. Chronic nicotine exposure alters blood-brain barrier permeability and diminishes brain uptake of methyllycaconitine. J. Neurochem. 2005, 94, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Rahman, S.; Cucullo, L. Drugs of abuse and blood-brain barrier endothelial dysfunction: A focus on the role of oxidative stress. J. Cereb. Blood Flow Metab. 2016, 36, 539–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.Q.; Wei, M.D.; Wang, K.W.; Lan, Y.X.; Zhu, N.; Wang, Y. Nicotine contributes to the neural stem cells fate against toxicity of microglial-derived factors induced by Abeta via the Wnt/beta-catenin pathway. Int. J. Neurosci. 2016, 126, 257–268. [Google Scholar] [CrossRef]
- Campos, M.W.; Serebrisky, D.; Castaldelli-Maia, J.M. Smoking and Cognition. Curr. Drug Abuse Rev. 2016, 9, 76–79. [Google Scholar] [CrossRef]
- Leventhal, A.M.; Waters, A.J.; Boyd, S.; Moolchan, E.T.; Lerman, C.; Pickworth, W.B. Gender differences in acute tobacco withdrawal: Effects on subjective, cognitive, and physiological measures. Exp. Clin. Psychopharmacol. 2007, 15, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Heishman, S.J.; Kleykamp, B.A.; Singleton, E.G. Meta-analysis of the acute effects of nicotine and smoking on human performance. Psychopharmacology 2010, 210, 453–469. [Google Scholar] [CrossRef] [Green Version]
- Hendricks, P.S.; Ditre, J.W.; Drobes, D.J.; Brandon, T.H. The early time course of smoking withdrawal effects. Psychopharmacology 2006, 187, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.R.; Keenan, R.M.; Yellin, A. Effect of tobacco withdrawal on sustained attention. Addict. Behav. 1989, 14, 577–580. [Google Scholar] [CrossRef]
- Jacobsen, L.K.; Mencl, W.E.; Constable, R.T.; Westerveld, M.; Pugh, K.R. Impact of smoking abstinence on working memory neurocircuitry in adolescent daily tobacco smokers. Psychopharmacology 2007, 193, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Mendrek, A.; Monterosso, J.; Simon, S.L.; Jarvik, M.; Brody, A.; Olmstead, R.; Domier, C.P.; Cohen, M.S.; Ernst, M.; London, E.D. Working memory in cigarette smokers: Comparison to non-smokers and effects of abstinence. Addict. Behav. 2006, 31, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, P.S.; Cobb, A.R.; Moissinac, L.; Hirshman, E. Evidence that episodic memory impairment during tobacco abstinence is independent of attentional mechanisms. J. Gen. Psychol. 2010, 137, 331–342. [Google Scholar] [CrossRef]
- Miech, R.; Patrick, M.E.; O’Malley, P.M.; Johnston, L.D. E-cigarette use as a predictor of cigarette smoking: Results from a 1-year follow-up of a national sample of 12th grade students. Tob. Control. 2017, 26, e106–e111. [Google Scholar] [CrossRef]
- Cullen, K.A.; Gentzke, A.S.; Sawdey, M.D.; Chang, J.T.; Anic, G.M.; Wang, T.W.; Creamer, M.R.; Jamal, A.; Ambrose, B.K.; King, B.A. e-Cigarette Use Among Youth in the United States, 2019. JAMA 2019, 322, 2095. [Google Scholar] [CrossRef]
- Willett, J.G.; Bennett, M.; Hair, E.C.; Xiao, H.; Greenberg, M.S.; Harvey, E.; Cantrell, J.; Vallone, D.M. Recognition, use and perceptions of JUUL among youth and young adults. Tob. Control. 2019, 28, 115–116. [Google Scholar] [CrossRef]
- Xu, C.; Loh, H.H.; Law, P.Y. Effects of addictive drugs on adult neural stem/progenitor cells. Cell Mol. Life Sci. 2016, 73, 327–348. [Google Scholar] [CrossRef]
- Lee, H.; Park, J.R.; Yang, J.; Kim, E.; Hong, S.H.; Woo, H.M.; Ryu, S.M.; Cho, S.J.; Park, S.M.; Yang, S.R. Nicotine inhibits the proliferation by upregulation of nitric oxide and increased HDAC1 in mouse neural stem cells. Vitr. Cell. Dev. Biol. Anim. 2014, 50, 731–739. [Google Scholar] [CrossRef]
- Helen, G.S.; Havel, C.; Dempsey, D.A.; Jacob, P.; Benowitz, N.L. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction 2016, 111, 535–544. [Google Scholar] [CrossRef]
- Widera, D.; Mikenberg, I.; Elvers, M.; Kaltschmidt, C.; Kaltschmidt, B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.H.; Bonde, S.; Kokaia, Z.; Jacobsen, S.-E.W.; Lindvall, O. Tumor Necrosis Factor Receptor 1 Is a Negative Regulator of Progenitor Proliferation in Adult Hippocampal Neurogenesis. J. Neurosci. 2006, 26, 9703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klassen, H.; Imfeld, K.L.; Kirov, I.I.; Tai, L.; Gage, F.H.; Young, M.J.; Berman, M.A. Expression of cytokines by multipotent neural progenitor cells. Cytokine 2003, 22, 101–106. [Google Scholar] [CrossRef]
- Volkow, N.D. Epigenetics of nicotine: Another nail in the coughing. Sci. Transl. Med. 2011, 3, 107ps43. [Google Scholar] [CrossRef] [Green Version]
- Pance, A.; Chantome, A.; Reveneau, S.; Bentrari, F.; Jeannin, J.F. A repressor in the proximal human inducible nitric oxide synthase promoter modulates transcriptional activation. FASEB J. 2002, 16, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yon, J.-M.; Jung, A.Y.; Lee, J.G.; Jung, K.Y.; Kang, J.-K.; Lee, B.J.; Yun, Y.W.; Nam, S.Y. Resveratrol prevents nicotine-induced teratogenesis in cultured mouse embryos. Reprod. Toxicol. 2012, 34, 340–346. [Google Scholar] [CrossRef]
- Wątroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef]
- Hou, Y.; Chen, H.; He, Q.; Jiang, W.; Luo, T.; Duan, J.; Mu, N.; He, Y.; Wang, H. Changes in methylation patterns of multiple genes from peripheral blood leucocytes of Alzheimer’s disease patients. Acta Neuropsychiatr. 2013, 25, 66–76. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, F.; Cheng, L.; Luo, T.; Xu, J.; Wang, H.-Q. Expression Profiles of SIRT1 and APP Genes in Human Neuroblastoma SK-N-SH Cells Treated with Two Epigenetic Agents. Neurosci. Bull. 2016, 32, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Abrous, D.N.; Adriani, W.; Montaron, M.-F.; Aurousseau, C.; Rougon, G.; Le Moal, M.; Piazza, P.V. Nicotine self-administration impairs hippocampal plasticity. J. Neurosci. 2002, 22, 3656–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scerri, C.; Stewart, C.A.; Breen, K.C.; Balfour, D.J.K. The effects of chronic nicotine on spatial learning and bromodeoxyuridine incorporation into the dentate gyrus of the rat. Psychopharmacology 2006, 184, 540–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shingo, A.S.; Kito, S. Effects of nicotine on neurogenesis and plasticity of hippocampal neurons. J Neural Transm. 2005, 112, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Arai, Y. Highly polysialylated neural cell adhesion molecule (NCAM-H) is expressed by newly generated granule cells in the dentate gyrus of the adult rat. J. Neurosci. 1993, 13, 2351–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremer, H.; Chazal, G.; Lledo, P.; Rougon, G.; Montaron, M.; Mayo, W.; Le Moal, M.; Abrous, D. PSA-NCAM: An important regulator of hippocampal plasticity. Int J Dev Neurosci 2000, 18, 213–220. [Google Scholar] [CrossRef]
- Levin, E.D. Nicotinic systems and cognitive function. Psychopharmacology 1992, 108, 417–431. [Google Scholar] [CrossRef]
- Levin, E.D.; Briggs, S.J.; Christopher, N.C.; Rose, J.E. Persistence of chronic nicotine-induced cognitive facilitation. Behav. Neural. Biol. 1992, 58, 152–158. [Google Scholar] [CrossRef]
- Levin, E.D.; Rezvani, A.H. Development of nicotinic drug therapy for cognitive disorders. Eur. J. Pharmacol. 2000, 393, 141–146. [Google Scholar] [CrossRef]
- Shingo, A.S.; Kito, S. Estrogen induces insulin-like growth factor-1 mRNA expression in the immortalized hippocampal cell: Determination by quantitative real-time polymerase chain reaction. Neurochem Res. 2003, 28, 1379–1383. [Google Scholar] [CrossRef]
- Mudo, G.; Belluardo, N.; Mauro, A.; Fuxe, K. Acute intermittent nicotine treatment induces fibroblast growth factor-2 in the subventricular zone of the adult rat brain and enhances neuronal precursor cell proliferation. Neuroscience 2007, 145, 470–483. [Google Scholar] [CrossRef]
- Cohen, A.; Soleiman, M.T.; Talia, R.; Koob, G.F.; George, O.; Mandyam, C.D. Extended access nicotine self-administration with periodic deprivation increases immature neurons in the hippocampus. Psychopharmacology 2015, 232, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, T. Hippocampal adult neurogenesis occurs in a microenvironment provided by PSA-NCAM-expressing immature neurons. J. Neurosci. Res. 2002, 69, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Snyder, F.R.; Davis, F.C.; Henningfield, J.E. The tobacco withdrawal syndrome: Performance decrements assessed on a computerized test battery. Drug Alcohol. Depend. 1989, 23, 259–266. [Google Scholar] [CrossRef]
- Hamidovic, A.; Candelaria, L.; Rodriguez, I.; Yamada, M.; Nawarskas, J.; Burge, M.R. Learning and memory performance following acute intranasal insulin administration in abstinent smokers. Hum. Psychopharmacol. 2018, 33, e2649. [Google Scholar] [CrossRef] [PubMed]
- Hirshman, E.; Rhodes, D.K.; Zinser, M.; Merritt, P. The effect of tobacco abstinence on recognition memory, digit span recall, and attentional vigilance. Exp. Clin. Psychopharmacol. 2004, 12, 76–83. [Google Scholar] [CrossRef]
- Myers, C.S.; Taylor, R.C.; Moolchan, E.T.; Heishman, S.J. Dose-related enhancement of mood and cognition in smokers administered nicotine nasal spray. Neuropsychopharmacology 2008, 33, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Campbell, N.R.; Fernandes, C.C.; Halff, A.W.; Berg, D.K. Endogenous signaling through alpha7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J. Neurosci. 2010, 30, 8734–8744. [Google Scholar] [CrossRef] [Green Version]
- Harrist, A.; Beech, R.D.; King, S.L.; Zanardi, A.; Cleary, M.A.; Caldarone, B.J.; Eisch, A.; Zoli, M.; Picciotto, M.R. Alteration of hippocampal cell proliferation in mice lacking the beta 2 subunit of the neuronal nicotinic acetylcholine receptor. Synapse 2004, 54, 200–206. [Google Scholar] [CrossRef]
- Mechawar, N.; Saghatelyan, A.; Grailhe, R.; Scoriels, L.; Gheusi, G.; Gabellec, M.-M.; Lledo, P.-M.; Changeux, J.-P. Nicotinic receptors regulate the survival of newborn neurons in the adult olfactory bulb. Proc. Natl. Acad. Sci. USA 2004, 101, 9822–9826. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.B.; Lee, S.H.; Chae, K.R.; Kim, C.K.; Hwang, D.Y.; Kim, B.G.; Jee, S.W.; Lee, S.H.; Sin, J.S.; Bae, C.J.; et al. Nicotine leads to improvements in behavioral impairment and an increase in the nicotine acetylcholine receptor in transgenic mice. Neurochem. Res. 2008, 33, 1783–1788. [Google Scholar] [CrossRef]
- Brown, D.; Ramlochansingh, C.; Manaye, K.F.; Tizabi, Y. Nicotine promotes survival of cells expressing amyloid precursor protein and presenilin: Implication for Alzheimer’s disease. Neurosci. Lett. 2013, 535, 57–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quik, M.; Parameswaran, N.; McCallum, S.E.; Bordia, T.; Bao, S.; McCormack, A.; Kim, A.; Tyndale, R.F.; Langston, J.; Di Monte, D.A. Chronic oral nicotine treatment protects against striatal degeneration in MPTP-treated primates. J. Neurochem. 2006, 98, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Mallela, A.; Chin, M.; McIntosh, J.M.; Perez, X.A.; Bordia, T. Nicotine-mediated improvement in L-dopa-induced dyskinesias in MPTP-lesioned monkeys is dependent on dopamine nerve terminal function. Neurobiol. Dis. 2013, 50, 30–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quik, M.; Bordia, T.; Zhang, D.; Perez, X.A. Nicotine and Nicotinic Receptor Drugs: Potential for Parkinson’s Disease and Drug-Induced Movement Disorders. Int. Rev. Neurobiol. 2015, 124, 247–271. [Google Scholar]
- Dong, J.; Cui, Y.; Li, S.; Le, W. Current Pharmaceutical Treatments and Alternative Therapies of Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 339–355. [Google Scholar] [CrossRef]
- Barreto, G.E.; Iarkov, A.; Moran, V.E. Beneficial effects of nicotine, cotinine and its metabolites as potential agents for Parkinson’s disease. Front Aging Neurosci. 2014, 6, 340. [Google Scholar]
- Inestrosa, N.C.; Godoy, J.A.; Vargas, J.Y.; Arrazola, M.S.; Rios, J.A.; Carvajal, F.J.; Serrano, F.G.; Farias, G.G. Nicotine prevents synaptic impairment induced by amyloid-beta oligomers through alpha7-nicotinic acetylcholine receptor activation. Neuromolecular Med. 2013, 15, 549–569. [Google Scholar] [CrossRef]
- Ren, Z.; Yang, M.; Guan, Z.; Yu, W. Astrocytic alpha7 Nicotinic Receptor Activation Inhibits Amyloid-beta Aggregation by Upregulating Endogenous alphaB-crystallin through the PI3K/Akt Signaling Pathway. Curr. Alzheimer Res. 2019, 16, 39–48. [Google Scholar] [CrossRef]
- Wei, P.; Liu, Q.; Li, D.; Zheng, Q.; Zhou, J.; Li, J. Acute nicotine treatment attenuates lipopolysaccharide-induced cognitive dysfunction by increasing BDNF expression and inhibiting neuroinflammation in the rat hippocampus. Neurosci. Lett. 2015, 604, 161–166. [Google Scholar] [CrossRef]
- Kalkman, H.O.; Feuerbach, D. Modulatory effects of α7 nAChRs on the immune system and its relevance for CNS disorders. Cell. Mol. Life Sci. 2016, 73, 2511–2530. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, K.; Jiang, D.; Wang, Y.; Xiao, X.; Zhu, N.; Li, M.; Jia, S.; Wang, Y. Blocking mPTP on Neural Stem Cells and Activating the Nicotinic Acetylcholine Receptor alpha7 Subunit on Microglia Attenuate Abeta-Induced Neurotoxicity on Neural Stem Cells. Neurochem. Res. 2016, 41, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Balducci, C. Alzheimer’s Disease, Oligomers, and Inflammation. J. Alzheimers Dis. 2018, 62, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozin, S.A.; Barykin, E.P.; Mitkevich, V.A.; Makarov, A.A. Anti-amyloid Therapy of Alzheimer’s Disease: Current State and Prospects. Biochemistry 2018, 83, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T.; et al. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Abeta accumulation through suppression of neuronal gamma-secretase activity and promotion of microglial amyloid-beta phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar]
- Ma, K.-G.; Qian, Y. Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 2019, 73, 96–106. [Google Scholar] [CrossRef]
- Sato, K. Effects of Microglia on Neurogenesis. Glia 2015, 63, 1394–1405. [Google Scholar] [CrossRef] [Green Version]
- Mondello, S.; Hayes, R.L. Chapter 16—Biomarkers, in Handbook of Clinical Neurology; Grafman, J., Salazar, A.M., Eds.; Elsevier: Cambridge, MA, USA, 2015; pp. 245–265. [Google Scholar] [CrossRef]
- Wainer, B.H.; Levey, A.I.; Mufson, E.J. Mesulam MM Chapter 5—Cholinergic systems in mammalian brain identified with antibodies against choline acetyltransferase. In Selected Topics from Neurochemistry; Osborne, N.N., Ed.; Amsterdam: Pergamon, Turkey, 1985; pp. 85–109. [Google Scholar]
- Stadler, J.O.; Bentz, B.G.; Harbrecht, B.G.; Di Silvio, M.; Curran, R.D.; Billiar, T.R.; Hoffman, R.A.; Simmons, R.L. Tumor necrosis factor alpha inhibits hepatocyte mitochondrial respiration. Ann. Surg. 1992, 216, 539–546. [Google Scholar] [CrossRef]
- Xie, Z.; Smith, C.J.; Van Eldik, L.J. Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia 2004, 45, 170–179. [Google Scholar] [CrossRef]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.-C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef]
- Samavati, L.; Lee, I.; Mathes, I.; Lottspeich, F.; Hüttemann, M. Tumor Necrosis Factor α Inhibits Oxidative Phosphorylation through Tyrosine Phosphorylation at Subunit I of Cytochrome c Oxidase. J. Biol. Chem. 2008, 283, 21134–21144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voloboueva, L.A.; Lee, S.W.; Emery, J.F.; Palmer, T.D.; Giffard, R. Mitochondrial Protection Attenuates Inflammation-Induced Impairment of Neurogenesis In Vitro and In Vivo. J. Neurosci. 2010, 30, 12242–12251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A. Biochemistry: A pore way to die. Nature 2005, 434, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Hirata, N.; Sekino, Y.; Kanda, Y. Role of α7-nicotinic acetylcholine receptor in normal and cancer stem cells. Curr. Drug Targets 2012, 13, 656–665. [Google Scholar]
- Narla, S.T.; Klejbor, I.; Birkaya, B.; Lee, Y.-W.; Moryś, J.; Stachowiak, E.K.; Prokop, D.; Bencherif, M.; Stachowiak, M.K. Activation of Developmental Nuclear Fibroblast Growth Factor Receptor 1 Signaling and Neurogenesis in Adult Brain by α7 Nicotinic Receptor Agonist. STEM CELLS Transl. Med. 2013, 2, 776–788. [Google Scholar] [CrossRef]
- Gao, J.; Sana, R.; Calder, V.; Calonge, M.; Lee, W.; Wheeler, L.A.; Stern, M.E. Mitochondrial Permeability Transition Pore in Inflammatory Apoptosis of Human Conjunctival Epithelial Cells and T Cells: Effect of Cyclosporin A. Investig. Opthalmology Vis. Sci. 2013, 54, 4717–4733. [Google Scholar] [CrossRef]
- Lisowski, P.; Kannan, P.; Mlody, B.G.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef]
- Berger, E.; Rath, E.; Yuan, D.; Waldschmitt, N.; Khaloian, S.; Allgäuer, M.; Staszewski, O.; Lobner, E.M.; Schöttl, T.; Giesbertz, P.; et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 2016, 7, 13171. [Google Scholar] [CrossRef] [Green Version]
- García-Prat, L.; Sousa-Victor, P.; Muñoz-Cánoves, P. Proteostatic and Metabolic Control of Stemness. Cell Stem Cell 2017, 20, 593–608. [Google Scholar] [CrossRef] [Green Version]
- Margineantu, D.H.; Hockenbery, D.M. Mitochondrial functions in stem cells. Curr. Opin. Genet. Dev. 2016, 38, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.J.; Yoon, S.H.; Do, J.T. Mitochondrial Dynamics in Stem Cells and Differentiation. Int. J. Mol. Sci. 2018, 19, 3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Duan, S.; Yi, F.; Ocampo, A.; Liu, G.H.; Belmonte, J.C. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013, 18, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.N.; Hartman, J.H.; Mello, D.F. Mitochondrial Toxicity. Toxicol. Sci. 2018, 162, 15–23. [Google Scholar] [CrossRef]
- Godoy, J.A.; Valdivieso, A.G.; Inestrosa, N.C. Nicotine Modulates Mitochondrial Dynamics in Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 8965–8977. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Prasad, S.K.; Banerjee, O.; Singh, S.; Banerjee, A.; Bose, A.; Pal, S.; Maji, B.K.; Mukherjee, S. Targeting mitochondria with folic acid and vitamin B12ameliorates nicotine mediated islet cell dysfunction. Environ. Toxicol. 2018, 33, 988–1000. [Google Scholar] [CrossRef]
- Malińska, D.; Więckowski, M.R.; Michalska, B.; Drabik, K.; Prill, M.; Patalas-Krawczyk, P.; Walczak, J.; Szymański, J.; Mathis, C.; Van Der Toorn, M.; et al. Mitochondria as a possible target for nicotine action. J. Bioenerg. Biomembr. 2019, 51, 259–276. [Google Scholar] [CrossRef] [Green Version]
- Zahedi, A.; Phandthong, R.; Chaili, A.; Leung, S.; Omaiye, E.; Talbot, P. Mitochondrial Stress Response in Neural Stem Cells Exposed to Electronic Cigarettes. iScience 2019, 16, 250–269. [Google Scholar] [CrossRef] [Green Version]
- Bahl, V.; Johnson, K.; Phandthong, R.; Zahedi, A.; Schick, S.F.; Talbot, P. From the Cover: Thirdhand Cigarette Smoke Causes Stress-Induced Mitochondrial Hyperfusion and Alters the Transcriptional Profile of Stem Cells. Toxicol. Sci. 2016, 153, 55–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Marín-Burgin, A.; Schinder, A.F. Requirement of adult-born neurons for hippocampus-dependent learning. Behav. Brain Res. 2012, 227, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhao, H.; Gleichman, A.; Machnicki, M.; Telang, S.; Tang, S.; Rshtouni, M.; Ruddell, J.H.; Carmichael, S.T. Region-specific and activity-dependent regulation of SVZ neurogenesis and recovery after stroke. Proc. Natl. Acad. Sci. USA 2019, 116, 13621–13630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, B.; Peron, S.; Murray, K.; Jaber, M.; Gaillard, A. Cortical lesion stimulates adult subventricular zone neural progenitor cell proliferation and migration to the site of injury. Stem Cell Res. 2013, 11, 965–977. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, S.; Goings, G.E.; Soderstrom, K.E.; Szele, F.G.; Kozlowski, D.A. Cellular proliferation and migration following a controlled cortical impact in the mouse. Brain Res. 2005, 1053, 38–53. [Google Scholar] [CrossRef]
- Salman, H.; Ghosh, P.; Kernie, S.G. Subventricular Zone Neural Stem Cells Remodel the Brain following Traumatic Injury in Adult Mice. J. Neurotrauma 2004, 21, 283–292. [Google Scholar] [CrossRef]
- Zhang, R.L.; Chopp, M.; Roberts, C.; Liu, X.; Wei, M.; Nejad-Davarani, S.P.; Wang, X.; Zhang, Z.G. Stroke Increases Neural Stem Cells and Angiogenesis in the Neurogenic Niche of the Adult Mouse. PLoS ONE 2014, 9, e113972. [Google Scholar] [CrossRef] [Green Version]
- Thored, P.; Wood, J.; Arvidsson, A.; Cammenga, J.; Kokaia, Z.; Lindvall, O. Long-Term Neuroblast Migration Along Blood Vessels in an Area With Transient Angiogenesis and Increased Vascularization After Stroke. Stroke 2007, 38, 3032–3039. [Google Scholar] [CrossRef] [Green Version]
- Faiz, M.; Acarin, L.; Villapol, S.; Schulz, S.; Castellano, B.; Gonzalez, B. Substantial migration of SVZ cells to the cortex results in the generation of new neurons in the excitotoxically damaged immature rat brain. Mol. Cell. Neurosci. 2008, 38, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Jaber, M.; Gaillard, A. Potentials of endogenous neural stem cells in cortical repair. Front. Cell. Neurosci. 2012, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.J.; Yu, S.; Lee, J.Y.; Hoffer, B.; Wang, Y. Improving Neurorepair in Stroke Brain Through Endogenous Neurogenesis-Enhancing Drugs. Cell Transplant. 2017, 26, 1596–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, S.-H.; Park, H.-H. Neurogenesis in Stroke Recovery. Transl. Stroke Res. 2017, 8, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-R.; Willing, A. Enhancing endogenous capacity to repair a stroke-damaged brain: An evolving field for stroke research. Prog. Neurobiol. 2018, 163, 5–26. [Google Scholar] [CrossRef]
- Benraiss, A.; Chmielnicki, E.; Lerner, K.; Roh, D.; Goldman, S.A. Adenoviral Brain-Derived Neurotrophic Factor Induces Both Neostriatal and Olfactory Neuronal Recruitment from Endogenous Progenitor Cells in the Adult Forebrain. J. Neurosci. 2001, 21, 6718–6731. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jin, K.; Mao, X.O.; Xie, L.; Banwait, S.; Marti, H.H.; Greenberg, D.A. VEGF-overexpressing transgenic mice show enhanced post-ischemic neurogenesis and neuromigration. J. Neurosci. Res. 2007, 85, 740–747. [Google Scholar] [CrossRef]
- Clark, A.R.; Carter, A.B.; Hager, L.E.; Price, E.M. In Vivo Neural Tissue Engineering: Cylindrical Biocompatible Hydrogels That Create New Neural Tracts in the Adult Mammalian Brain. Stem Cells Dev. 2016, 25, 1109–1118. [Google Scholar] [CrossRef]
- Rodier, P.M. Developing brain as a target of toxicity. Environ. Health Perspect. 1995, 103 (Suppl. 6), 73–76. [Google Scholar]
- Pericot-Valverde, I.; Gaalema, D.E.; Priest, J.S.; Higgins, S.T. E-cigarette awareness, perceived harmfulness, and ever use among U.S. adults. Prev. Med. 2017, 104, 92–99. [Google Scholar] [CrossRef]
- Berger, F.; Bedrosian, T.A.; Vijayaraghavan, S. Nicotinic Receptor-Induced Apoptotic Cell Death of Hippocampal Progenitor Cells. J. Neurosci. 1998, 18, 6871–6881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joschko, M.A.; Dreosti, I.E.; Tulsi, R.S. The teratogenic effects of nicotine in vitro in rats: A light and electron microscope study. Neurotoxicology Teratol. 1991, 13, 307–316. [Google Scholar] [CrossRef]
- Slotkin, T.A. If nicotine is a developmental neurotoxicant in animal studies, dare we recommend nicotine replacement therapy in pregnant women and adolescents? Neurotoxicol. Teratol. 2008, 30, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.B.; Broide, R.S.; Leslie, F.M. Nicotine and brain development. Birth Defects Res. Part C: Embryo Today: Rev. 2008, 84, 30–44. [Google Scholar] [CrossRef]
- Liu, F.; Tao, X.; Pang, G.; Wu, D.; Hu, Y.; Xue, S.; Liu, J.; Li, B.; Zhou, L.; Liu, Q.; et al. Maternal Nicotine Exposure During Gestation and Lactation Period Affects Behavior and Hippocampal Neurogenesis in Mouse Offspring. Front. Pharmacol. 2020, 10, 1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoli, M.; Le Novere, N.; Hill, J.A.; Changeux, J.P. Developmental regulation of nicotinic ACh receptor subunit mRNAs in the rat central and peripheral nervous systems. J. Neurosci. 1995, 15, 1912–1939. [Google Scholar] [CrossRef] [PubMed]
- Tribollet, E.; Bertrand, D.; Marguerat, A.; Raggenbass, M. Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: An autoradiographic study in the rat brain. Neuroscience 2004, 124, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Pomeroy, S.L. 131-Development of the Nervous System, in Fetal and Neonatal Physiology, 5th ed.; Polin, R.A., Abman, S.H., Rowitch, D.H., Benitz, W.E., Fox, W.W., Eds.; Elsevier: Cambridge, MA, USA, 2017; pp. 1294–1313. [Google Scholar]
- Naeff, B.; Schlumpf, M.; Lichtensteiger, W. Pre- and postnatal development of high-affinity [3H]nicotine binding sites in rat brain regions: An autoradiographic study. Dev. Brain Res. 1992, 68, 163–174. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, C.; Miao, H.; Gong, Z.H.; Nordberg, A. Postnatal changes of nicotinic acetylcholine receptor alpha 2, alpha 3, alpha 4, alpha 7 and beta 2 subunits genes expression in rat brain. Int. J. Dev. Neurosci. 1998, 16, 507–518. [Google Scholar] [CrossRef]
- Hohmann, C.F. A morphogenetic role for acetylcholine in mouse cerebral neocortex. Neurosci. Biobehav. Rev. 2003, 27, 351–363. [Google Scholar] [CrossRef]
- Bryden, D.W.; Burton, A.C.; Barnett, B.R.; Cohen, V.J.; Hearn, T.N.; Jones, E.A.A.; Kariyil, R.J.; Kunin, A.; Kwak, S.I.; Lee, J.; et al. Prenatal Nicotine Exposure Impairs Executive Control Signals in Medial Prefrontal Cortex. Neuropsychopharmacology 2016, 41, 716–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, J.B.; McQuown, S.C.; Leslie, F.M. The dynamic effects of nicotine on the developing brain. Pharmacol. Ther. 2009, 122, 125–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskenazi, B.; Castorina, R. Association of prenatal maternal or postnatal child environmental tobacco smoke exposure and neurodevelopmental and behavioral problems in children. Environ. Health Perspect. 1999, 107, 991–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, M.; Moolchan, E.T.; Robinson, M.L. Behavioral and Neural Consequences of Prenatal Exposure to Nicotine. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Mojica, C.; Bai, Y.; Lotfipour, S. Maternal nicotine exposure effects on adolescent learning and memory are abolished in alpha(α)2* nicotinic acetylcholine receptor-null mutant mice. Neuropharmacology 2018, 135, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Hu, W.; Zhou, X.; Lin, H.; Lan, L.; Wang, P.; Lv, W.; Geng, Q.; Xia, Y. Prenatal exposure to maternal smoking during pregnancy and attention-deficit/hyperactivity disorder in offspring: A meta-analysis. Reprod. Toxicol. 2018, 76, 63–70. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, W.; He, G.; Zhang, D.; Zhao, X.; Dai, J.; Wu, J.; Cao, Y.; Wang, Z.; Wang, L.; et al. Maternal nicotine exposure has severe cross-generational effects on offspring behavior. Behav. Brain Res. 2018, 348, 263–266. [Google Scholar] [CrossRef]
- McGrath-Morrow, S.A.; Gorzkowski, J.; Groner, J.A.; Rule, A.M.; Wilson, K.; Tanski, S.E.; Collaco, J.M.; Klein, J.D. The Effects of Nicotine on Development. Pediatrics 2020, 145, e20191346. [Google Scholar] [CrossRef]
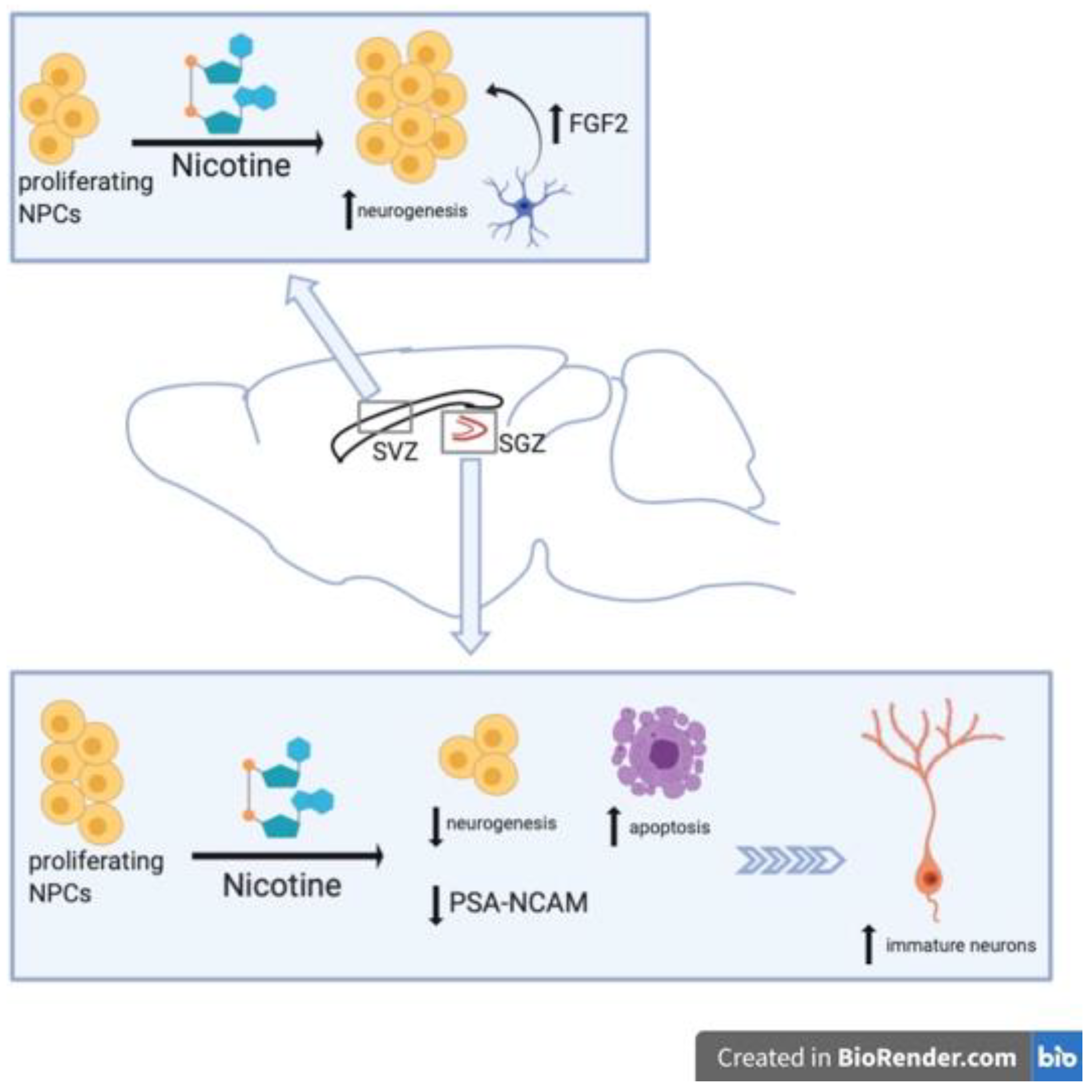
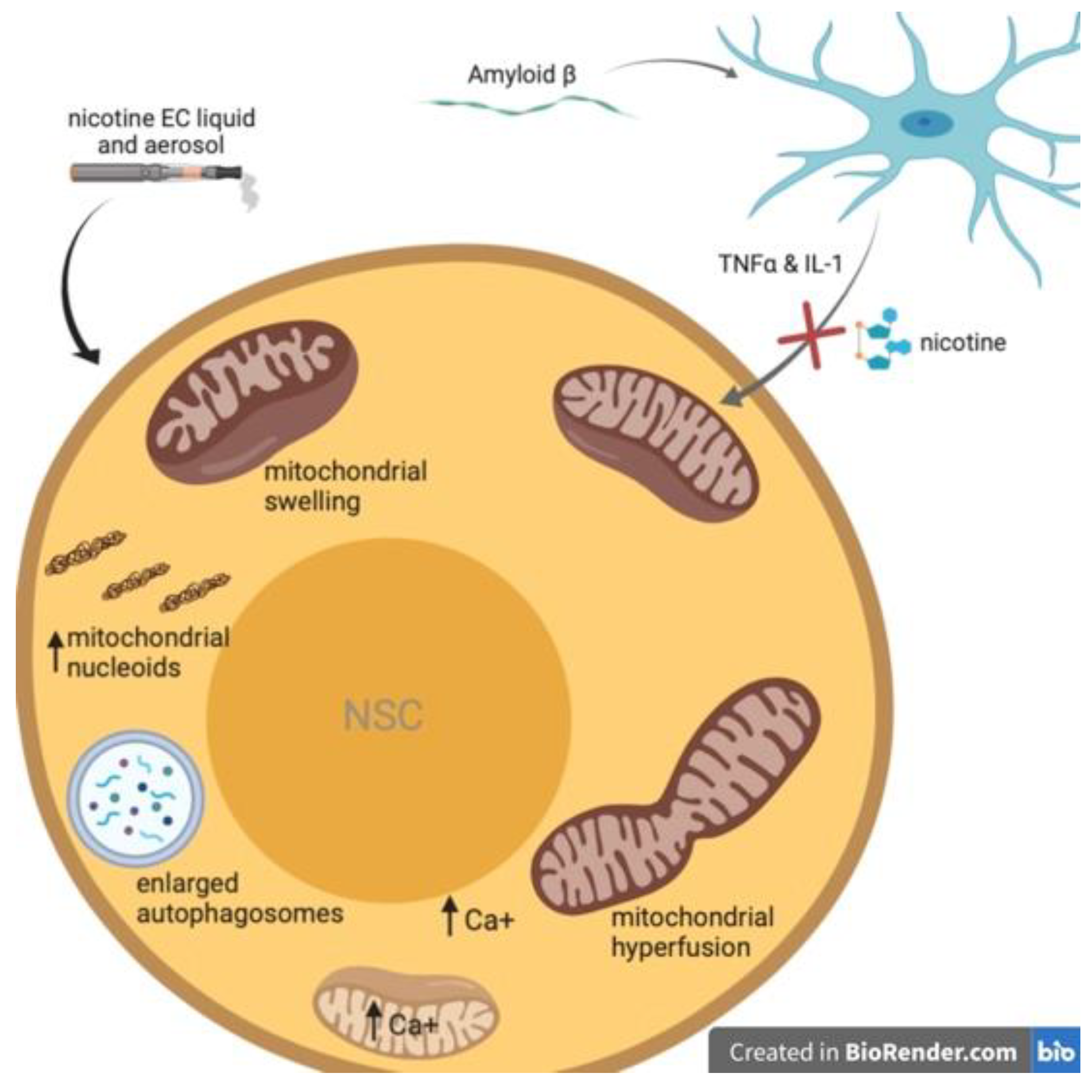

{kind=link}
{kind=link}
| Observation | Nicotine Doses | Model System | Ref |
|---|---|---|---|
| Nicotine reduces proliferation of NSCs via transcriptional mechanisms | 100, 400, 800 µM | Neurospheres | [40] |
| Nicotine reduced the NSC proliferation in the dentate gyrus (DG) | 4 mg/kg/day | Mice | [52] |
| Nicotine reduced the NSC proliferation in the DG | Variable (IVSA) | Rats | [55] |
| Nicotine decreases hippocampal neurogenesis | 0.1, 0.5, or 1 mg/kg | Rats | [53] |
| Observation | Nicotine Doses | Model System | Ref |
|---|---|---|---|
| Intermittent nicotine exposure increases neural precursor proliferation in the SVZ due to increases in FGF-2 | 1 mg/kg | Rats | [60] |
| Nicotine dependence and deprivation increases the number of immature neurons in the SGZ of the DG | Variable, IVSA. | Rats | [61] |
| Observation | Nicotine Doses | Model System | Ref |
|---|---|---|---|
| Nicotine prevents Aβ aggregation via hippocampal α7 nAChRs | 10 µM (cultures), 1 mg/kg (mice) | Cultured hippocampal neurons, mice | [77] |
| Nicotine is neuroprotective by reducing inflammation by activating BDNF/TrkB and inhibiting TNF-α and IL-1 | 0.5 mg/kg | Rats | [79] |
| Nicotine-induced activation of α7 nAChRs attenuates Aβ-induced neurotoxicity by reducing Aβ accumulation in mitochondria | 1 µM | cultured hippocampal NSCs | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brooks, A.C.; Henderson, B.J. Systematic Review of Nicotine Exposure’s Effects on Neural Stem and Progenitor Cells. Brain Sci. 2021, 11, 172. https://doi.org/10.3390/brainsci11020172
Brooks AC, Henderson BJ. Systematic Review of Nicotine Exposure’s Effects on Neural Stem and Progenitor Cells. Brain Sciences. 2021; 11(2):172. https://doi.org/10.3390/brainsci11020172
Chicago/Turabian StyleBrooks, Arrin C., and Brandon J. Henderson. 2021. "Systematic Review of Nicotine Exposure’s Effects on Neural Stem and Progenitor Cells" Brain Sciences 11, no. 2: 172. https://doi.org/10.3390/brainsci11020172