
EEG Patterns in Patients with Prader–Willi Syndrome


 ,
,  ,
,  ,
,  and
and  on behalf of Genetic Obesity Study Group of the Italian Society of Pediatric Endocrinology and Diabetology (ISPED)
on behalf of Genetic Obesity Study Group of the Italian Society of Pediatric Endocrinology and Diabetology (ISPED)
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Patients Features
3.2. EEG Findings
3.3. Brain MRI Findings
3.4. Phenotype and Genotype Characteristics of PWS Patients with EEG Paroxysmal Abnormalities Versus PWS Patients without EEG Paroxysmal Abnormalities
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi Syndrome—Clinical Genetics, Diagnosis and Treatment Approaches: An Update. Curr. Pediatr. Rev. 2019, 15, 207–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.J.; Hou, J.W.; Sue, W.C.; Lee, W.T. Electroclinical characteristics of seizures-comparing Prader-Willi syndrome with Angelman syndrome. Brain Dev. 2005, 27, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kumada, T.; Ito, M.; Miyajima, T.; Fujii, T.; Okuno, T.; Go, T.; Hattori, H.; Yoshioka, M.; Kobayashi, K.; Kanazawa, O.; et al. Multi-institutional study on the correlation between chromosomal abnormalities and epilepsy. Brain Dev. 2005, 27, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Greenwood, R.; Fisher, A.; Pendyal, S.; Powell, C.M. Characteristics and frequency of seizure disorder in 56 patients with Prader-Willi syndrome. Am. J. Med. Genet. A 2009, 149, 1581–1584. [Google Scholar] [CrossRef] [PubMed]
- Vendrame, M.; Maski, K.P.; Chatterjee, M.; Heshmati, A.; Krishnamoorthy, K.; Tan, W.H.; Kothare, S.V. Epilepsy in Prader-Willi syndrome: Clinical characteristics and correlation to genotype. Epilepsy Behav. 2010, 19, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, E.; Murakami, N.; Sakuta, R.; Nagai, T. Evaluating the frequency and characteristics of seizures in 142 Japanese patients with Prader-Willi syndrome. Am. J. Med. Genet. A 2013, 161, 2052–2055. [Google Scholar] [CrossRef]
- Verrotti, A.; Cusmai, R.; Laino, D.; Carotenuto, M.; Esposito, M.; Falsaperla, R.; Margari, L.; Rizzo, R.; Savasta, S.; Grosso, S.; et al. Long-term outcome of epilepsy in patients with Prader-Willi syndrome. J. Neurol. 2015, 262, 116–123. [Google Scholar] [CrossRef]
- Elia, M. Chromosomal abnormalities and cortical malformations. In Clinical Electroencephalography; Mecarelli, O., Ed.; Springer: Cham, Switzerland, 2019; pp. 547–585. [Google Scholar]
- Varela, M.C.; Kok, F.; Setian, N.; Kim, C.A.; Koiffmann, C.P. Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: Study of 75 patients. Clin. Genet. 2005, 67, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Sinnema, M.; Maaskant, M.A.; van Schrojenstein Lantman-de Valk, H.M.; van Nieuwpoort, I.C.; Drent, M.L.; Curfs, L.M.; Schrander-Stumpel, C.T. Physical health problems in adults with Prader-Willi syndrome. Am. J. Med. Genet. A 2011, 155, 2112–2124. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, T.; Gross-Tsur, V. Epilepsy in Prader-Willi syndrome: Experience of a national referral centre. Dev. Med. Child Neurol. 2013, 55, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Holm, V.A.; Cassidy, S.B.; Butler, M.G.; Hanchett, J.M.; Greenswag, L.R.; Whitman, B.Y.; Greenberg, F. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics 1993, 91, 398–402. [Google Scholar]
- American Society of Human Genetics/American College of Medical Genetics. Diagnostic testing for Prader-Willi and Angelman syndromes: Report of the ASHG/ACMG test and technology transfer committee. Am. J. Hum. Genet. 1996, 58, 1085–1088. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Gardner, R.J.; Crossland, K.M.; Scheffer, I.E.; Berkovic, S.F. Chromosomal abnormalities and epilepsy: A review for clinicians and gene hunters. Epilepsia 2002, 43, 127–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavazzuti, G.B.; Cappella, L.; Nalin, A. Longitudinal study of epileptiform EEG patterns in normal children. Epilepsia 1980, 21, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Pelc, K.; Boyd, S.G.; Cheron, G.; Dan, B. Epilepsy in Angelman syndrome. Seizure 2008, 17, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Iughetti, L.; Bosio, L.; Corrias, A.; Gargantini, L.; Ragusa, L.; Livieri, C.; Predieri, B.; Bruzzi, P.; Caselli, G.; Grugni, G. Pituitary height and neuroradiological alterations in patients with Prader-Labhart-Willi syndrome. Eur. J. Pediatr. 2008, 167, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Grugni, G.; Crinò, A.; De Bellis, A.; Convertino, A.; Bocchini, S.; Maestrini, S.; Cirillo, P.; De Lucia, S.; Delvecchio, M.; on behalf of the Italian Autoimmune Hypophysitis Network Study and of the Genetic Obesity Study Group of the Italian Society of Pediatric Endocrinology and Diabetology (ISPED). Autoimmune pituitary involvement in Prader Willi syndrome: New perspective for further research. Endocrine 2018, 62, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Goldstone, A.P.; Couch, J.A.; Shuster, J.; He, G.; Driscoll, D.J.; Liu, Y.; Schmalfuss, I.M. Pituitary abnormalities in Prader-Willi syndrome and early onset morbid obesity. Am. J. Med. Genet. A 2008, 146, 570–577. [Google Scholar] [CrossRef] [PubMed]
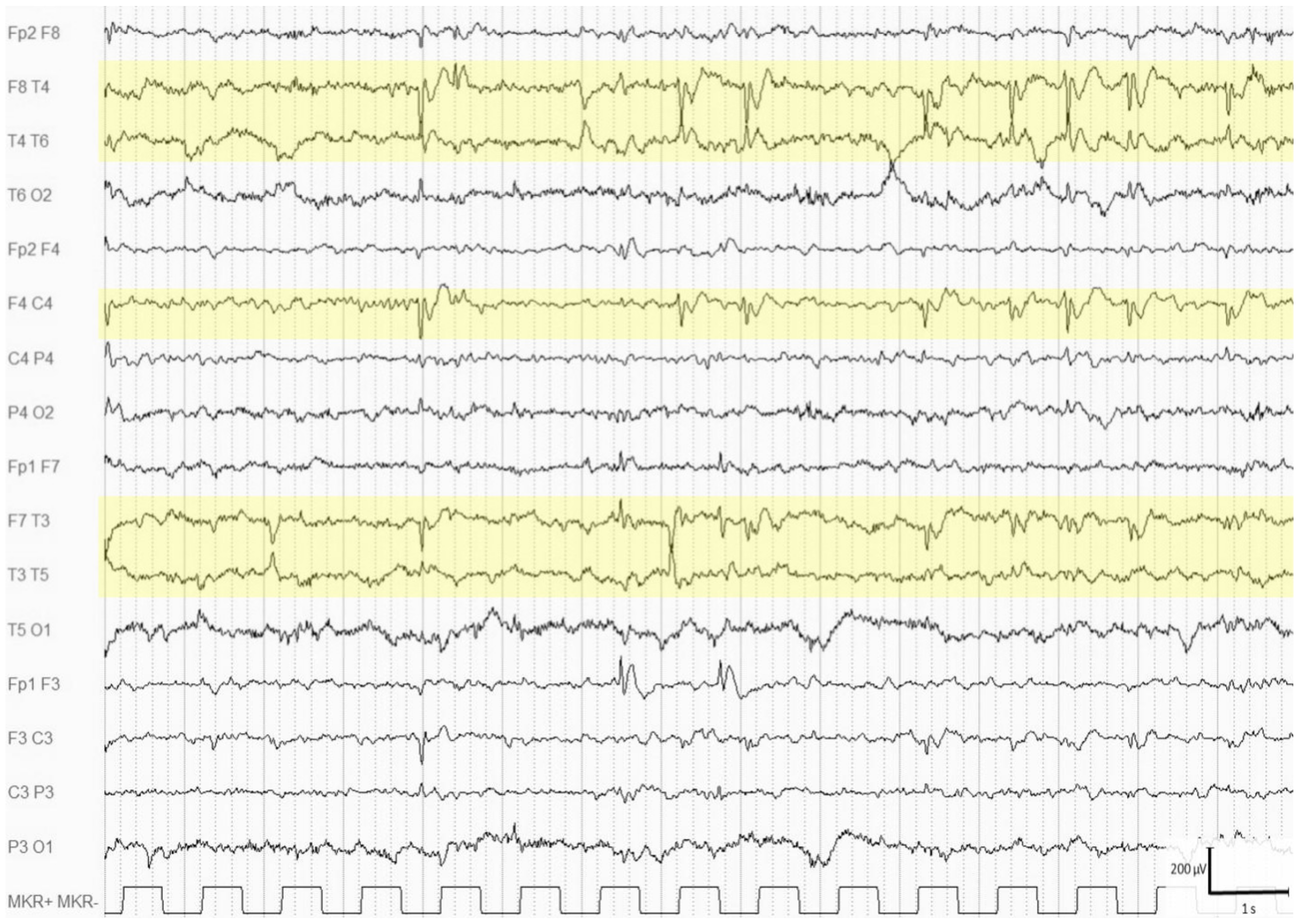



{kind=link}
{kind=link}
{kind=link}
| Variables | Results |
|---|---|
| Gender (males/female) | 37/35 (52.7%/47.3%) |
| Age range (years) | 2–42 |
| Follow-up duration (years) | 2.9 ± 3.9 (0.1–19.0) |
| Genotype | |
| 15q11-q13 paternal deletion Maternal UPD ICD | 32 (43.2%) 38 (51.4%) 4 (5.4%) |
| Intellectual disability Mild Moderate Severe | 67 (90.5%) 47 (63.5%) 15 (20.3%) 5 (6.7%) |
| BIF | 3 (4.1%) |
| Behavioral disturbances | 7 (9.5%) |
| Seizures Absences Focal seizures, impaired awareness GTCS | 4 (5.4%) 2 1 1 |
| Sleep apnea | 17 (23.0%) |
| Drug therapy | 59 (79.7%) |
| Variables | Results |
|---|---|
| Number of recorded EEGs | 198 (1–10) |
| Mean number of recorded EEGs ± SD | 2.9 ± 1.9 |
| Age at the first EEG (years) | 7.7 ± 7.8 (0.1–33) |
| Age at the last EEG (years) | 10.6 ± 8.3 (0.1–33) |
| EEG type | |
| Wakefulness Wakefulness and sleep | 92 (46.5%) 106 (53.5%) |
| Background activity at the last EEG | |
| Slow Normal | 0 (0%) 74 (100%) |
| Abnormalities Middle-anterior spikes Middle-posterior spikes Focal slow waves | 19 (25.7%) |
| 5 (26.3%) | |
| 12 (63.2%) | |
| 2 (10.5%) |
| Findings | Number (Rate) |
|---|---|
| Abnormal brain MRI | 23 (59%) * |
| Myelination anomalies | 4 (17.4%) |
| Corpus callosum hypoplasia | 3 (13%) |
| Pituitary hypoplasia | 9 (39.1%) |
| Subaracnoid space enlargement | 7 (30.4%) |
| Enlargement of the lateral ventricles | 9 (39.1%) |
| Arachnoid cyst | 5 (21.7%) |
| Cerebral atrophy | 5 (21.7%) |
| Cerebellar hypoplasia | 2 (8.7%) |
| PWS EEG+ Patients | PWS EEG- Patients | |
|---|---|---|
| Mean age (years) | 11.2 (range 3–33) | 10.3 (range 0.1–33) |
| Behavioral disturbances | 5 | 12 |
| Deletion 15q11-q13 | 10 | 22 |
| UPD | 9 | 29 |
| ICD | 0 | 4 |
| Brain abnormalities | 6 | 17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elia, M.; Rutigliano, I.; Sacco, M.; Madeo, S.F.; Wasniewska, M.; Li Pomi, A.; Trifirò, G.; Di Bella, P.; De Lucia, S.; Vetri, L.; et al. EEG Patterns in Patients with Prader–Willi Syndrome. Brain Sci. 2021, 11, 1045. https://doi.org/10.3390/brainsci11081045
Elia M, Rutigliano I, Sacco M, Madeo SF, Wasniewska M, Li Pomi A, Trifirò G, Di Bella P, De Lucia S, Vetri L, et al. EEG Patterns in Patients with Prader–Willi Syndrome. Brain Sciences. 2021; 11(8):1045. https://doi.org/10.3390/brainsci11081045
Chicago/Turabian StyleElia, Maurizio, Irene Rutigliano, Michele Sacco, Simona F. Madeo, Malgorzata Wasniewska, Alessandra Li Pomi, Giuliana Trifirò, Paolo Di Bella, Silvana De Lucia, Luigi Vetri, and et al. 2021. "EEG Patterns in Patients with Prader–Willi Syndrome" Brain Sciences 11, no. 8: 1045. https://doi.org/10.3390/brainsci11081045
APA StyleElia, M., Rutigliano, I., Sacco, M., Madeo, S. F., Wasniewska, M., Li Pomi, A., Trifirò, G., Di Bella, P., De Lucia, S., Vetri, L., Iughetti, L., & Delvecchio, M., on behalf of Genetic Obesity Study Group of the Italian Society of Pediatric Endocrinology and Diabetology (ISPED). (2021). EEG Patterns in Patients with Prader–Willi Syndrome. Brain Sciences, 11(8), 1045. https://doi.org/10.3390/brainsci11081045