
Effects of Delivering Guanidinoacetic Acid or Its Prodrug to the Neural Tissue: Possible Relevance for Creatine Transporter Deficiency

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Creatine and Its Procurement by the Organism
1.2. Use of Guanidinoacetate, the Creatine Precursor, as a Dietary Supplement
1.3. Possible Use of Guanidinoacetic Acid or of Its Prodrugs in Treating Creatine Transporter Deficiency
1.4. Aims of This Paper
2. Materials and Methods
2.1. Preparation of Hippocampal Slices
2.2. Inactivation of Creatine Transporter in Hippocampal Slices
2.3. Study on the Effect of GPA on Slices Viability
- ACSF
- ACSF + GPA (0.5 mM)
- ACSF + GPA (1 mM)
- ACSF + GPA (1 mM) + Diacetyl-GAAE (0, 1 mM)
2.4. Tissue Processing for Biochemical Experiments
- ACSF
- ACSF Cl-free
- ACSF + Creatine (2 mM)
- ACSF Cl-free + Creatine (2 mM)
- ACSF Cl-free + GAA (2 mM)
- ACSF Cl-free + Diacetyl-GAAE (0, 1 mM)
2.5. Synthesis of N-Diacetyl Guanidine Acetic Ethyl Ester (Diacetyl-GAAE)
2.5.1. Synthesis of Methyl N,N’-Diacetylcarbamimidothioate
2.5.2. Synthesis of (Z)-Ethyl 2-(2,3-Diacetylguanidino)acetate
2.6. HPLC and Mass Spectrometry Analysis
2.7. Measurement of Tissue Content of Creatine
3. Results
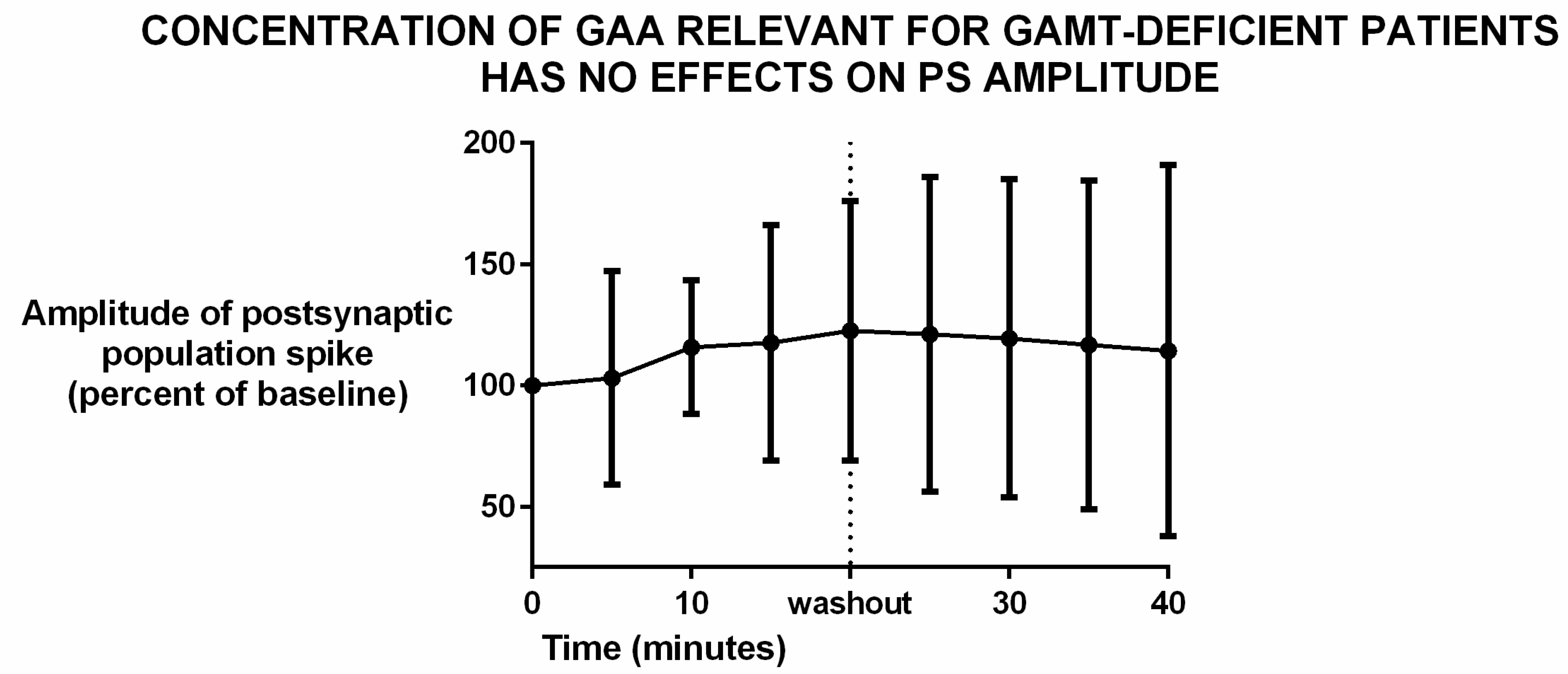
3.1. Effects of Guanidinoacetic Acid
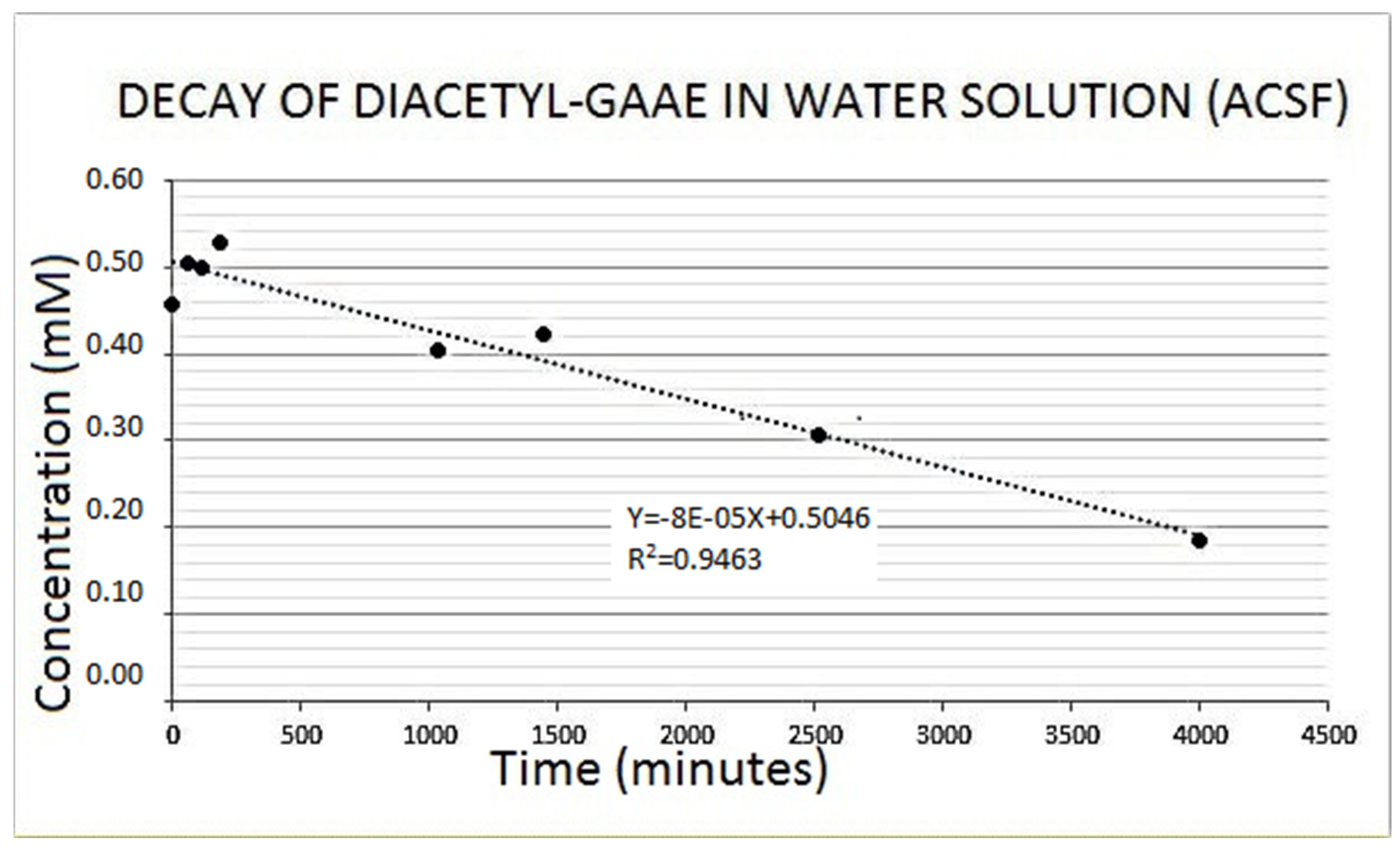
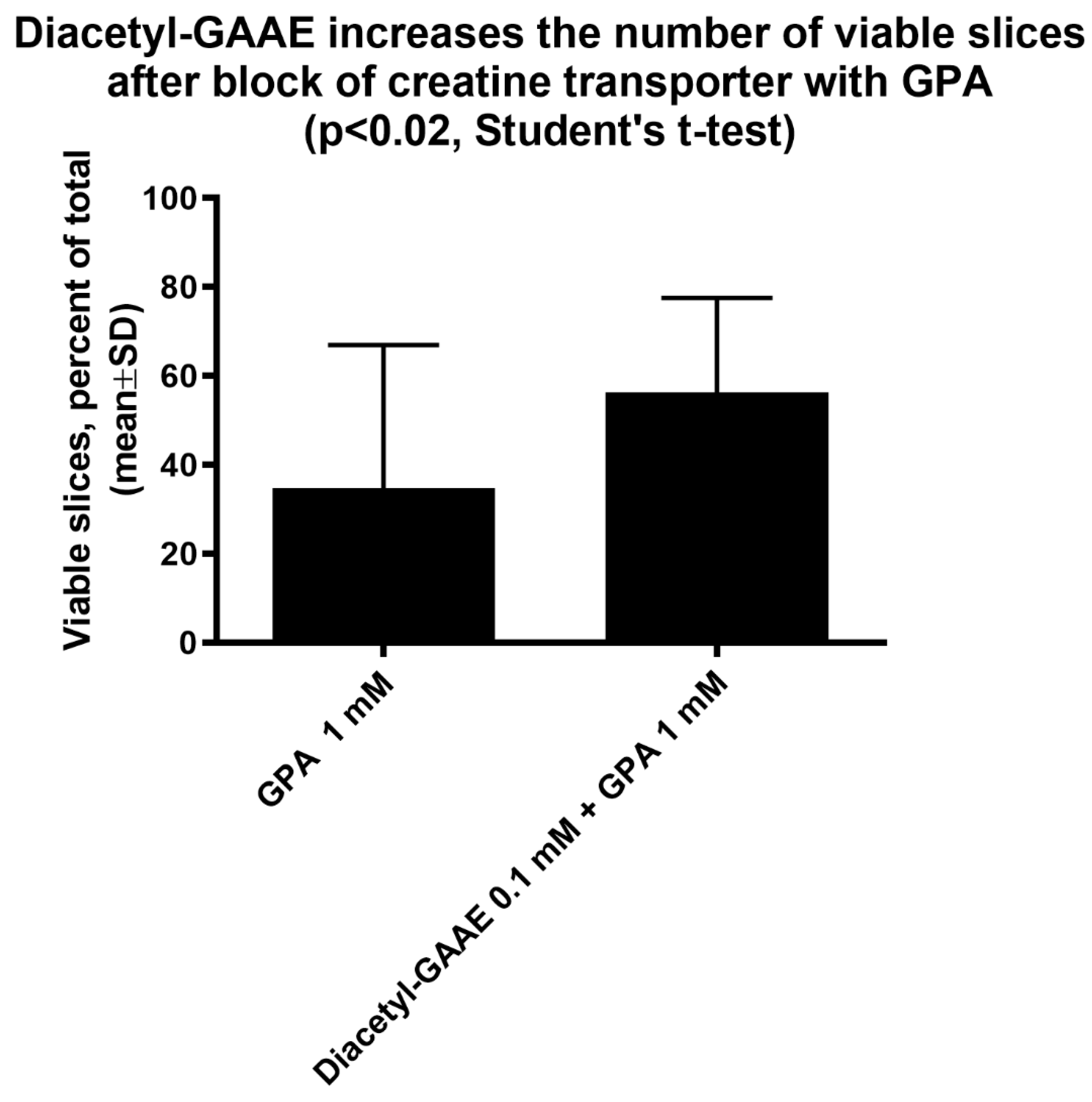
3.2. Diacetyl-Guanidinic Acid Ethyl Ester (Diacetyl-GAAE)
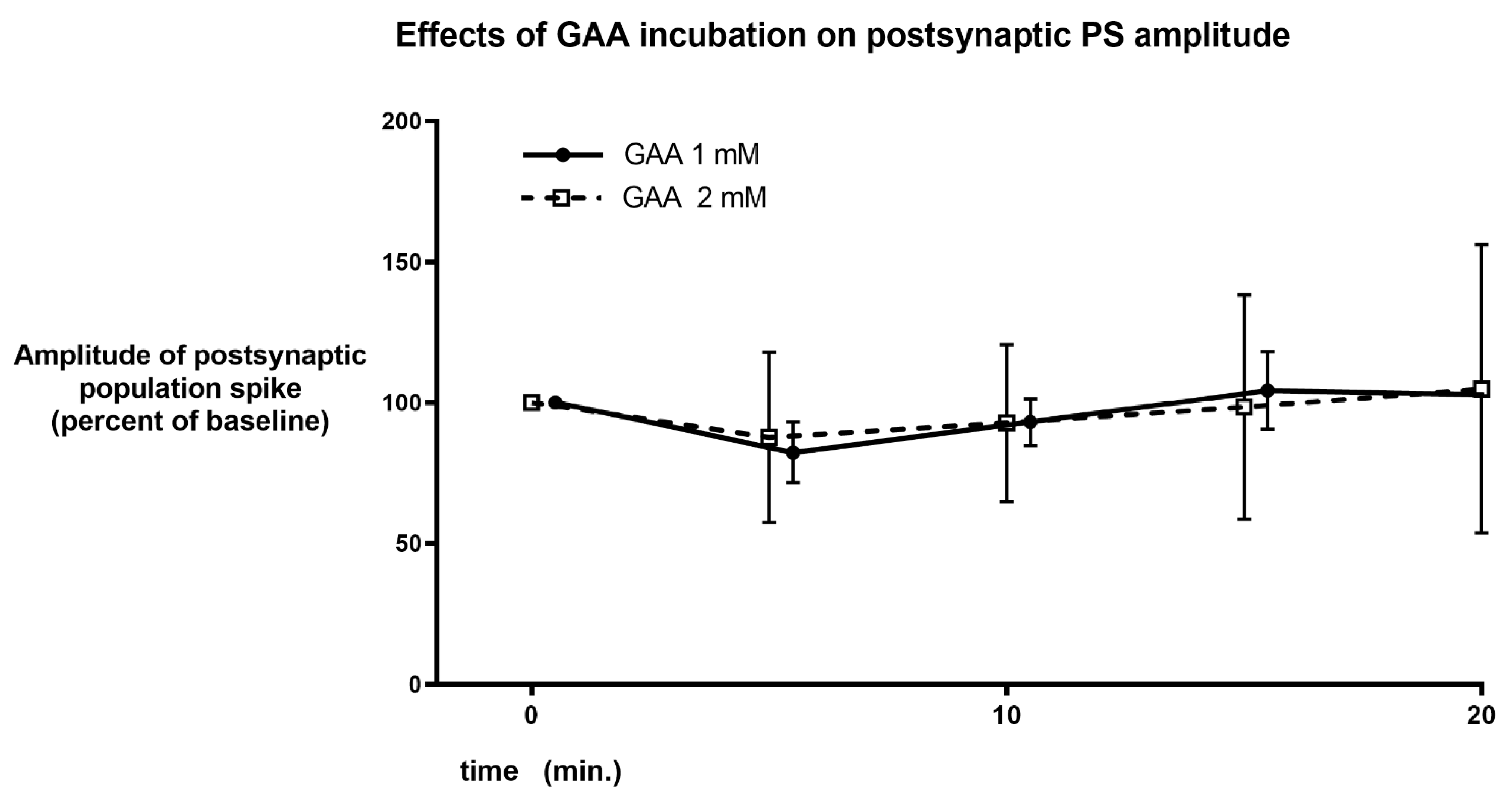
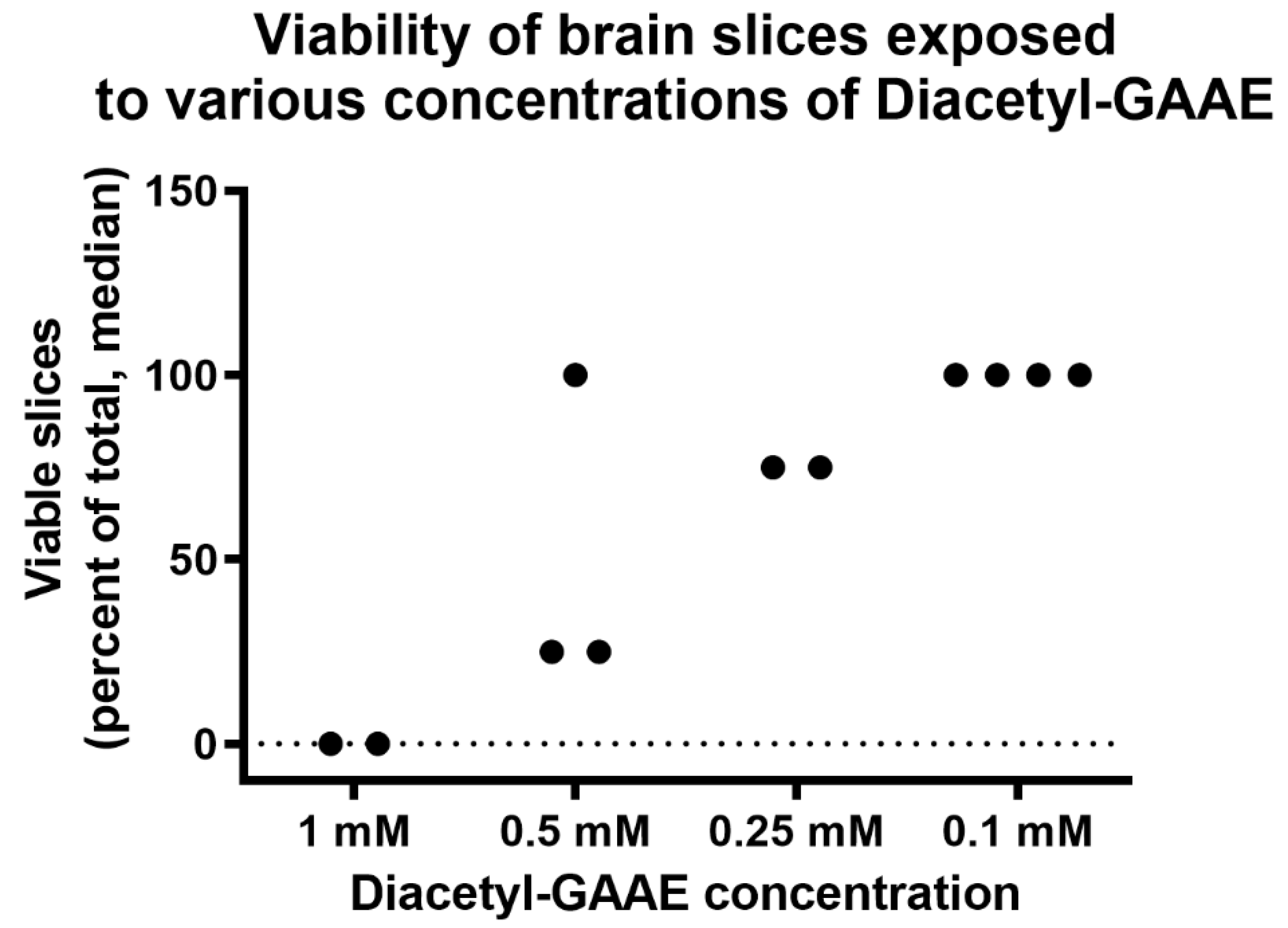
3.3. Effects of Diacetyl-GAAE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Greenhaff, P.L. The Creatine-Phosphocreatine System: There’s More than One Song in Its Repertoire. J. Physiol. 2001, 537, 657. [Google Scholar] [CrossRef]
- Adhihetty, P.J.; Beal, M.F. Creatine and Its Potential Therapeutic Value for Targeting Cellular Energy Impairment in Neurodegenerative Diseases. Neuromol. Med. 2008, 10, 275–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The Creatine Kinase System and Pleiotropic Effects of Creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef] [Green Version]
- Stockler-Ipsiroglu, S.; van Karnebeek, C.D.M. Cerebral Creatine Deficiencies: A Group of Treatable Intellectual Developmental Disorders. Semin. Neurol. 2014, 34, 350–356. [Google Scholar] [CrossRef]
- Hanna-El-Daher, L.; Braissant, O. Creatine Synthesis and Exchanges between Brain Cells: What Can Be Learned from Human Creatine Deficiencies and Various Experimental Models? Amino Acids 2016, 48, 1877–1895. [Google Scholar] [CrossRef]
- Kreider, R.B.; Kalman, D.S.; Antonio, J.; Ziegenfuss, T.N.; Wildman, R.; Collins, R.; Candow, D.G.; Kleiner, S.M.; Almada, A.L.; Lopez, H.L. International Society of Sports Nutrition Position Stand: Safety and Efficacy of Creatine Supplementation in Exercise, Sport, and Medicine. J. Int. Soc. Sports Nutr. 2017, 14, 18. [Google Scholar] [CrossRef]
- Balestrino, M.; Adriano, E. Beyond Sports: Efficacy and Safety of Creatine Supplementation in Pathological or Paraphysiological Conditions of Brain and Muscle. Med. Res. Rev. 2019, 39, 2427–2459. [Google Scholar] [CrossRef] [PubMed]
- Ostojic, S.M.; Drid, P.; Ostojic, J. Guanidinoacetic Acid Increases Skeletal Muscle Creatine Stores in Healthy Men. Nutr. Burbank Los Angel. Cty. Calif 2016, 32, 723–724. [Google Scholar] [CrossRef] [PubMed]
- Ostojic, S.M.; Ostojic, J.; Drid, P.; Vranes, M. Guanidinoacetic Acid versus Creatine for Improved Brain and Muscle Creatine Levels: A Superiority Pilot Trial in Healthy Men. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab. 2016, 41, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Schütz, P.W.; Stöckler, S. Creatine Deficiency Syndromes. In Neurobiology of Disease; Gilman, S., Ed.; Academic Press: Cambridge, MA, USA, 2007; pp. 33–41. [Google Scholar]
- Hanna-El-Daher, L.; Béard, E.; Henry, H.; Tenenbaum, L.; Braissant, O. Mild Guanidinoacetate Increase under Partial Guanidinoacetate Methyltransferase Deficiency Strongly Affects Brain Cell Development. Neurobiol. Dis. 2015, 79, 14–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrino, M.; Adriano, E. Presence of Guanidinoacetate May Compensate Creatine Absence and Account for Less Statin-Induced Muscle Damage in GAMT-Deficient Compared to AGAT-Deficient Mice. Amino Acids 2020, 52, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Ostojic, S.M.; Ostojic, J.; Drid, P.; Vranes, M.; Jovanov, P. Dietary Guanidinoacetic Acid Increases Brain Creatine Levels in Healthy Men. Nutr. Burbank Los Angel. Cty. Calif 2017, 33, 149–156. [Google Scholar] [CrossRef]
- Van de Kamp, J.M.; Mancini, G.M.; Salomons, G.S. X-Linked Creatine Transporter Deficiency: Clinical Aspects and Pathophysiology. J. Inherit. Metab. Dis. 2014, 37, 715–733. [Google Scholar] [CrossRef]
- Braissant, O. Creatine and Guanidinoacetate Transport at Blood-Brain and Blood-Cerebrospinal Fluid Barriers. J. Inherit. Metab. Dis. 2012, 35, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Trotier-Faurion, A.; Dézard, S.; Taran, F.; Valayannopoulos, V.; de Lonlay, P.; Mabondzo, A. Synthesis and Biological Evaluation of New Creatine Fatty Esters Revealed Dodecyl Creatine Ester as a Promising Drug Candidate for the Treatment of the Creatine Transporter Deficiency. J. Med. Chem. 2013, 56, 5173–5181. [Google Scholar] [CrossRef]
- Adriano, E.; Gulino, M.; Arkel, M.; Salis, A.; Damonte, G.; Liessi, N.; Millo, E.; Garbati, P.; Balestrino, M. Di-Acetyl Creatine Ethyl Ester, a New Creatine Derivative for the Possible Treatment of Creatine Transporter Deficiency. Neurosci. Lett. 2018, 665, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, G.; Parodi, A.; Perasso, L.; Pohvozcheva, A.V.; Scarrone, S.; Adriano, E.; Florio, T.; Gandolfo, C.; Cupello, A.; Burov, S.V.; et al. The Creatine Transporter Mediates the Uptake of Creatine by Brain Tissue, but Not the Uptake of Two Creatine-Derived Compounds. Neuroscience 2006, 142, 991–997. [Google Scholar] [CrossRef]
- Adriano, E.; Garbati, P.; Salis, A.; Damonte, G.; Millo, E.; Balestrino, M. Creatine Salts Provide Neuroprotection Even after Partial Impairment of the Creatine Transporter. Neuroscience 2017, 340, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Whittingham, T.S.; Lipton, P. Cerebral Synaptic Transmission During Anoxia Is Protected by Creatine. J. Neurochem. 1981, 37, 1618–1621. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Caldeira Araújo, H.; Smit, W.; Verhoeven, N.M.; Salomons, G.S.; Silva, S.; Vasconcelos, R.; Tomás, H.; Tavares de Almeida, I.; Jakobs, C.; Duran, M. Guanidinoacetate Methyltransferase Deficiency Identified in Adults and a Child with Mental Retardation. Am. J. Med. Genet. A 2005, 133, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Hosoya, K.-I. Transport Characteristics of Guanidino Compounds at the Blood-Brain Barrier and Blood-Cerebrospinal Fluid Barrier: Relevance to Neural Disorders. Fluids Barriers CNS 2011, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Garbati, P.; Adriano, E.; Salis, A.; Ravera, S.; Damonte, G.; Millo, E.; Balestrino, M. Effects of Amide Creatine Derivatives in Brain Hippocampal Slices, and Their Possible Usefulness for Curing Creatine Transporter Deficiency. Neurochem. Res. 2014, 39, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Melani, R.; Rebaudo, R.; Noraberg, J.; Zimmer, J.; Balestrino, M. Changes in Extracellular Action Potential Detect Kainic Acid and Trimethyltin Toxicity in Hippocampal Slice Preparations Earlier than Do MAP2 Density Measurements. Altern. Lab. Anim. ATLA 2005, 33, 379–386. [Google Scholar] [CrossRef]
- Furling, D.; Ghribi, O.; Lahsaini, A.; Mirault, M.E.; Massicotte, G. Impairment of Synaptic Transmission by Transient Hypoxia in Hippocampal Slices: Improved Recovery in Glutathione Peroxidase Transgenic Mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4351–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, A.; Ebinger, F.; Rating, D.; Mayatepek, E. Improving Treatment of Guanidinoacetate Methyltransferase Deficiency: Reduction of Guanidinoacetic Acid in Body Fluids by Arginine Restriction and Ornithine Supplementation. Mol. Genet. Metab. 2001, 74, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Viau, K.S.; Ernst, S.L.; Pasquali, M.; Botto, L.D.; Hedlund, G.; Longo, N. Evidence-Based Treatment of Guanidinoacetate Methyltransferase (GAMT) Deficiency. Mol. Genet. Metab. 2013, 110, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Zugno, A.I.; Franzon, R.; Chiarani, F.; Bavaresco, C.S.; Wannmacher, C.M.D.; Wajner, M.; Wyse, A.T.S. Evaluation of the Mechanism Underlying the Inhibitory Effect of Guanidinoacetate on Brain Na+, K+-ATPase Activity. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2004, 22, 191–196. [Google Scholar] [CrossRef]
- Andersen, P.; Bliss, T.V.P.; Skrede, K.K. Unit Analysis of Hippocampal Population Spikes. Exp. Brain Res. 1971, 13, 208–221. [Google Scholar] [CrossRef]
- Margineanu, D.G. Epileptic Hypersynchrony Revisited. Neuroreport 2010, 21, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, G. Seizures and Epilepsy. In Principles of Neural Science; Kandel, E.R., Jessel, T.M., Schwartz, J.H., Eds.; McGraw-Hill: New York, NY, USA, 1991. [Google Scholar]
- Köhling, R.; Melani, R.; Koch, U.; Speckmann, E.-J.; Koudelka-Hep, M.; Thiébaud, P.; Balestrino, M. Detection of Electrophysiological Indicators of Neurotoxicity in Human and Rat Brain Slices by a Three-Dimensional Microelectrode Array. Altern. Lab. Anim. ATLA 2005, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.; Stoppini, L.; Balestrino, M.; Eskes, C.; Griesinger, C.; Sobanski, T.; Whelan, M.; Hartung, T.; Coecke, S. Electrophysiological Recording of Re-Aggregating Brain Cell Cultures on Multi-Electrode Arrays to Detect Acute Neurotoxic Effects. Neurotoxicology 2007, 28, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Bausch, S.B. Axonal Sprouting of GABAergic Interneurons in Temporal Lobe Epilepsy. Epilepsy Behav. EB 2005, 7, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Kan, H.E.; Renema, W.K.J.; Isbrandt, D.; Heerschap, A. Phosphorylated Guanidinoacetate Partly Compensates for the Lack of Phosphocreatine in Skeletal Muscle of Mice Lacking Guanidinoacetate Methyltransferase. J. Physiol. 2004, 560, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, Y.; Degrauw, T.J.; Lindquist, D.M.; Blanco, V.M.; Pyne-Geithman, G.J.; Daikoku, T.; Chambers, J.B.; Benoit, S.C.; Clark, J.F. Cyclocreatine Treatment Improves Cognition in Mice with Creatine Transporter Deficiency. J. Clin. Investig. 2012, 122, 2837–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enrico, A.; Patrizia, G.; Luisa, P.; Alessandro, P.; Gianluigi, L.; Carlo, G.; Maurizio, B. Electrophysiology and Biochemical Analysis of Cyclocreatine Uptake and Effect in Hippocampal Slices. J. Integr. Neurosci. 2013, 12, 285–297. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adriano, E.; Salis, A.; Damonte, G.; Millo, E.; Balestrino, M. Effects of Delivering Guanidinoacetic Acid or Its Prodrug to the Neural Tissue: Possible Relevance for Creatine Transporter Deficiency. Brain Sci. 2022, 12, 85. https://doi.org/10.3390/brainsci12010085
Adriano E, Salis A, Damonte G, Millo E, Balestrino M. Effects of Delivering Guanidinoacetic Acid or Its Prodrug to the Neural Tissue: Possible Relevance for Creatine Transporter Deficiency. Brain Sciences. 2022; 12(1):85. https://doi.org/10.3390/brainsci12010085
Chicago/Turabian StyleAdriano, Enrico, Annalisa Salis, Gianluca Damonte, Enrico Millo, and Maurizio Balestrino. 2022. "Effects of Delivering Guanidinoacetic Acid or Its Prodrug to the Neural Tissue: Possible Relevance for Creatine Transporter Deficiency" Brain Sciences 12, no. 1: 85. https://doi.org/10.3390/brainsci12010085
APA StyleAdriano, E., Salis, A., Damonte, G., Millo, E., & Balestrino, M. (2022). Effects of Delivering Guanidinoacetic Acid or Its Prodrug to the Neural Tissue: Possible Relevance for Creatine Transporter Deficiency. Brain Sciences, 12(1), 85. https://doi.org/10.3390/brainsci12010085