
Imaging Clinical Subtypes and Associated Brain Networks in Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
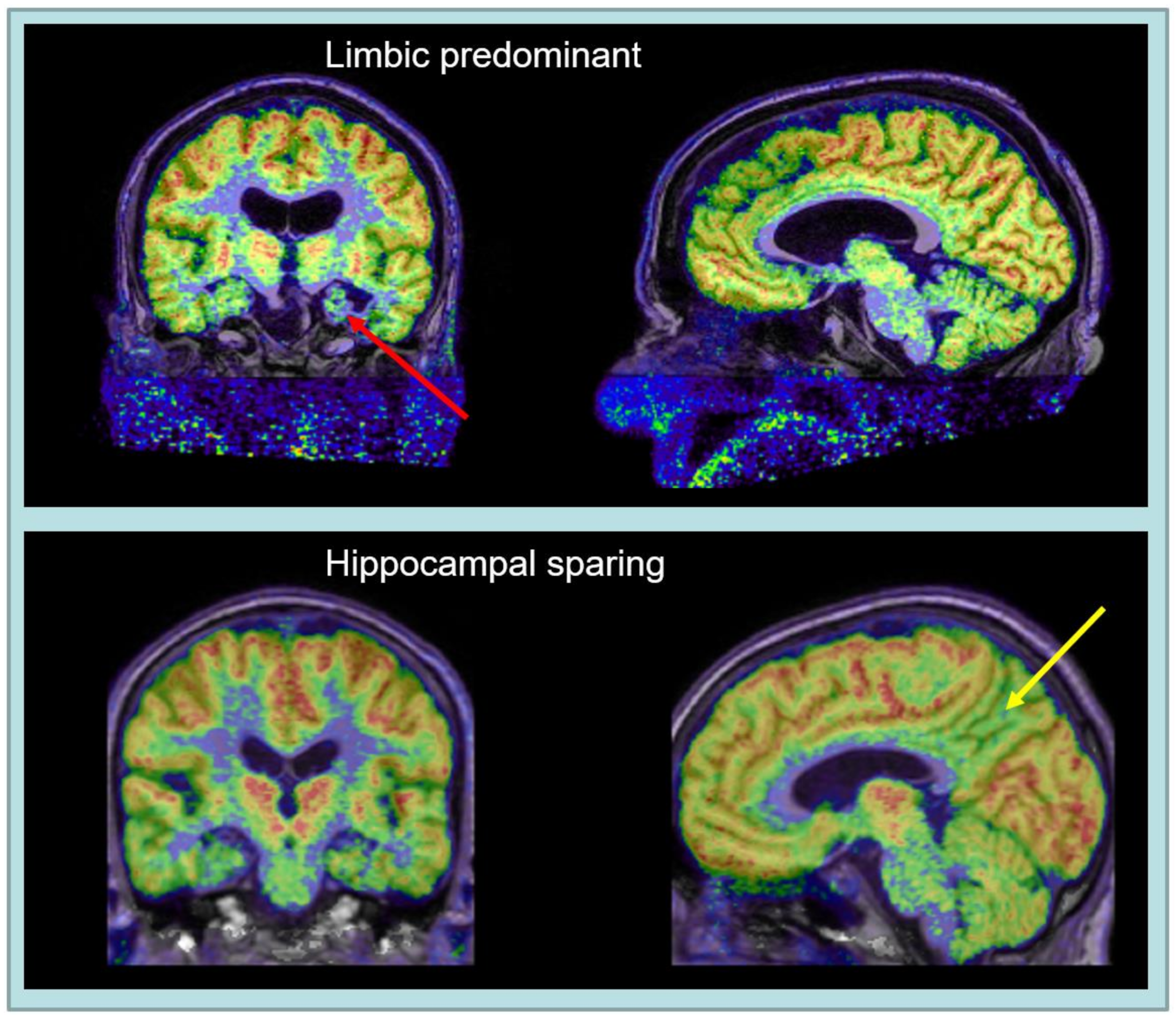
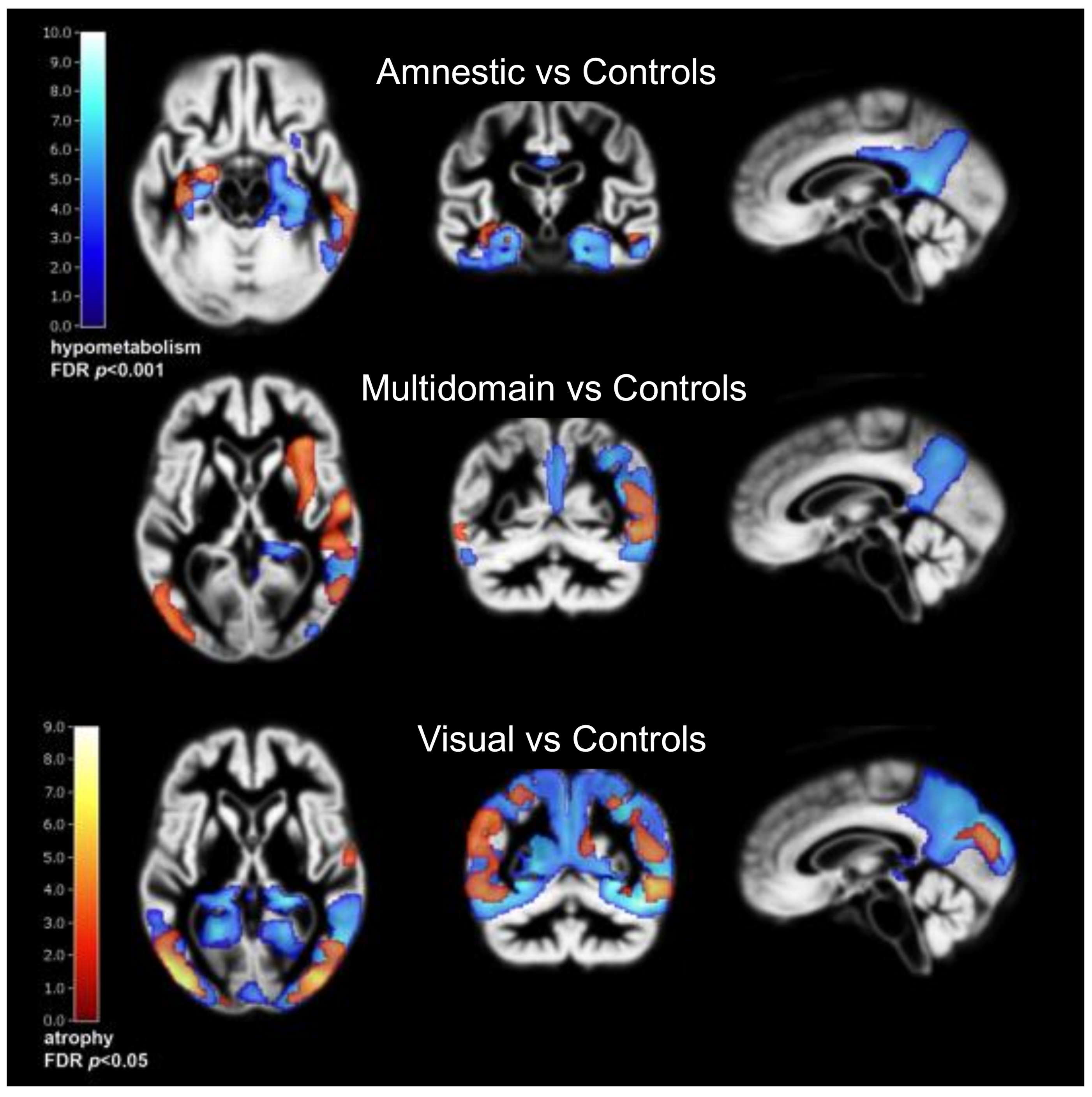
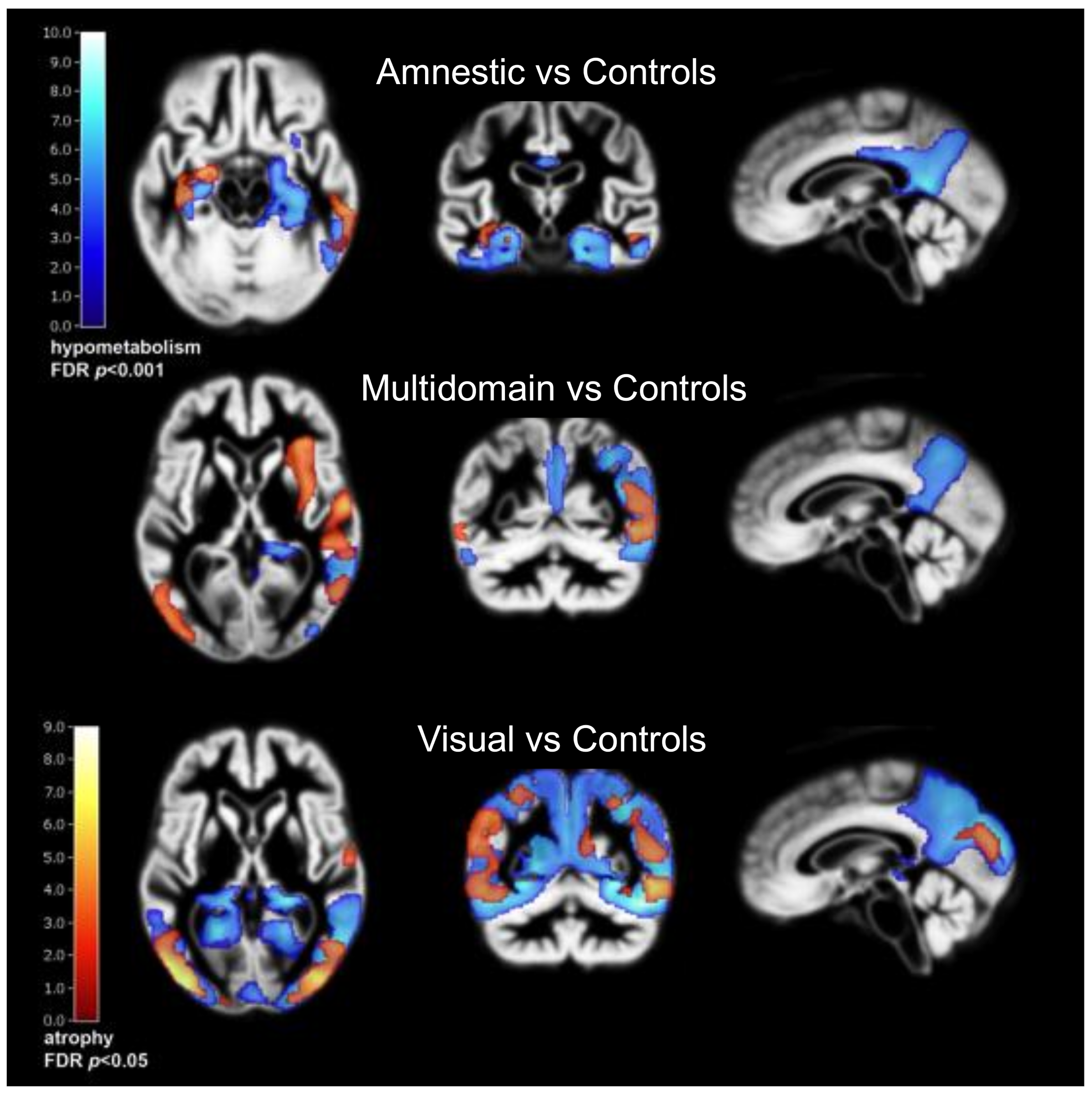
2. Alzheimer’s Disease Subtypes
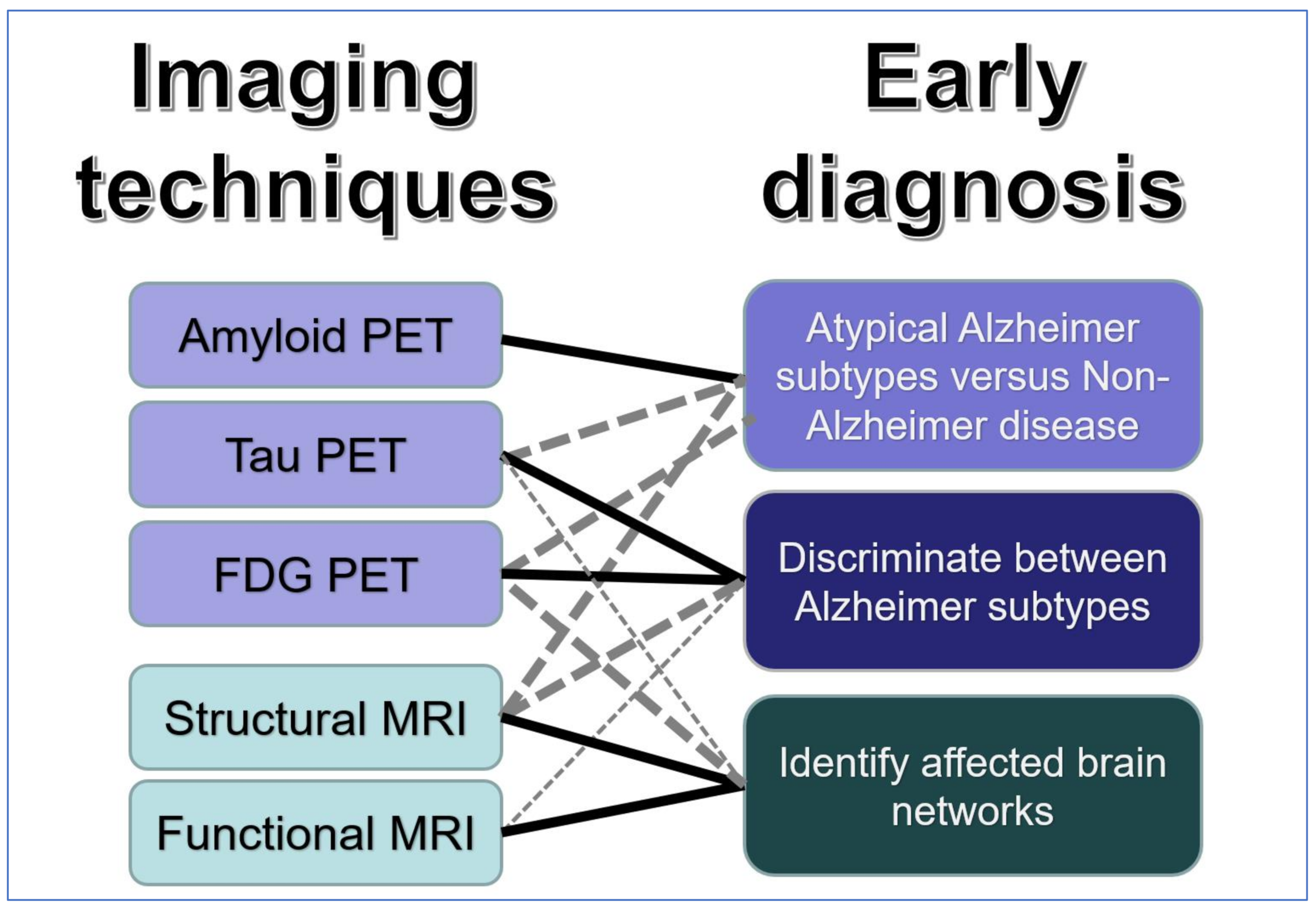
3. Imaging Techniques
3.1. Amyloid PET
3.2. Tau PET
3.3. FDG PET
3.4. Structural MRI
3.5. Functional MRI and Network Analysis
3.6. Neurotransmitter and Neuroinflammation Imaging
4. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hampel, H.; O’Bryant, S.E.; Durrleman, S.; Younesi, E.; Rojkova, K.; Escott-Price, V.; Corvol, J.-C.; Broich, K.; Dubois, B.; Lista, S.; et al. A Precision Medicine Initiative for Alzheimer’s disease: The road ahead to biomarker-guided integrative disease modeling. Climacteric 2017, 20, 107–118. [Google Scholar] [CrossRef]
- Ferrari, C.; Sorbi, S. The complexity of Alzheimer’s disease: An evolving puzzle. Physiol. Rev. 2021, 101, 1047–1081. [Google Scholar] [CrossRef]
- Ritchie, K.; Touchon, J. Heterogeneity in senile dementia of the Alzheimer type: Individual differences, progressive deterioration or clinical sub-types? J. Clin. Epidemiol. 1992, 45, 1391–1398. [Google Scholar] [CrossRef]
- Cummings, J.L. Cognitive and behavioral heterogeneity in Alzheimer’s disease: Seeking the neurobiological basis. Neurobiol. Aging 2000, 21, 845–861. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crutch, S.J.; Schott, J.M.; Rabinovici, G.D.; Murray, M.; Snowden, J.S.; van der Flier, W.M.; Dickerson, B.C.; Vandenberghe, R.; Ahmed, S.; Bak, T.H.; et al. Consensus classification of posterior cortical atrophy. Alzheimer’s Dement. 2017, 13, 870–884. [Google Scholar] [CrossRef]
- Johnson, J.K.; Head, E.; Kim, R.; Starr, A.; Cotman, C.W. Clinical and Pathological Evidence for a Frontal Variant of Alzheimer Disease. Arch. Neurol. 1999, 56, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, R. 179 Clinical subtypes of Alzheimer’s disease. Neurobiol. Aging 1996, 17, S45–S46. [Google Scholar] [CrossRef]
- Warren, J.D.; Fletcher, P.D.; Golden, H.L. The paradox of syndromic diversity in Alzheimer disease. Nat. Rev. Neurol. 2012, 8, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, D.; Nordberg, A.; Westman, E. Author Response: Biological Subtypes of Alzheimer Disease: A Systematic Review and Meta-analysis. Neurology 2021, 96, 238. [Google Scholar] [PubMed]
- Phillips, J.S.; Da Re, F.; Dratch, L.; Xie, S.X.; Irwin, D.J.; McMillan, C.T.; Vaishnavi, S.N.; Ferrarese, C.; Lee, E.B.; Shaw, L.M.; et al. Neocortical origin and progression of gray matter atrophy in nonamnestic Alzheimer’s disease. Neurobiol. Aging 2018, 63, 75–87. [Google Scholar] [CrossRef]
- Tang, M.; Ryman, D.C.; McDade, E.; Jasielec, M.S.; Buckles, V.D.; Cairns, N.J.; Fagan, A.M.; Goate, A.; Marcus, D.S.; Xiong, C.; et al. Neurological manifestations of autosomal dominant familial Alzheimer’s disease: A comparison of the published literature with the Dominantly Inherited Alzheimer Network observational study (DIAN-OBS). Lancet Neurol. 2016, 15, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Miller, Z.; Rosenberg, L.; Santos-Santos, M.A.; Stephens, M.; Allen, I.E.; Hubbard, H.I.; Cantwell, A.; Mandelli, M.L.; Grinberg, L.T.; Seeley, W.W.; et al. Prevalence of Mathematical and Visuospatial Learning Disabilities in Patients With Posterior Cortical Atrophy. JAMA Neurol. 2018, 75, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Miller, Z.; Mandelli, M.L.; Rankin, K.P.; Henry, M.; Babiak, M.C.; Frazier, D.T.; Lobach, I.V.; Bettcher, B.M.; Wu, T.Q.; Rabinovici, G.D.; et al. Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain 2013, 136, 3461–3473. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.H.C.; Ingelsson, M.; Watts, J.C. The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol. 2020, 142, 17–39. [Google Scholar] [CrossRef]
- Harris, J.M.; Gall, C.; Thompson, J.C.; Richardson, A.M.; Neary, D.; du Plessis, D.; Pal, P.; Mann, D.M.; Snowden, J.S.; Jones, M. Classification and pathology of primary progressive aphasia. Neurology 2013, 81, 1832–1839. [Google Scholar] [CrossRef]
- Nucera, A.; Hachinski, V. Cerebrovascular and Alzheimer disease: Fellow travelers or partners in crime? J. Neurochem. 2018, 144, 513–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habes, M.; Grothe, M.J.; Tunc, B.; McMillan, C.; Wolk, D.A.; Davatzikos, C. Disentangling Heterogeneity in Alzheimer’s Disease and Related Dementias Using Data-Driven Methods. Biol. Psychiatry 2020, 88, 70–82. [Google Scholar] [CrossRef]
- Mohanty, R.; Mårtensson, G.; Poulakis, K.; Muehlboeck, J.-S.; Rodriguez-Vieitez, E.; Chiotis, K.; Grothe, M.J.; Nordberg, A.; Ferreira, F.; Westman, E. Towards Harmonizing Subtyping Methods for Neuroimaging Studies in Alzheimer’s Disease [Internet]. April 2020, p. 2020.04.19.20064881. Available online: https://www.medrxiv.org/content/10.1101/2020.04.19.20064881v1 (accessed on 12 November 2021).
- Petersen, R.C.; Caracciolo, B.; Brayne, C.; Gauthier, S.; Jelic, V.; Fratiglioni, L. Mild cognitive impairment: A concept in evolution. J. Intern. Med. 2014, 275, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Whitwell, J.L.; Dickson, D.W.; Murray, M.E.; Weigand, S.D.; Tosakulwong, N.; Senjem, M.L.; Knopman, D.S.; Boeve, B.F.; Parisi, J.E.; Petersen, R.C.; et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer’s disease: A case-control study. Lancet Neurol. 2012, 11, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.M.; Pontecorvo, M.J.; Beach, T.G.; Bedell, B.J.; Coleman, R.E.; Doraiswamy, P.M.; Fleisher, A.S.; Reiman, E.M.; Sabbagh, M.N.; Sadowsky, C.H.; et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: A prospective cohort study. Lancet Neurol. 2012, 11, 669–678. [Google Scholar] [CrossRef]
- Sabri, O.; Sabbagh, M.N.; Seibyl, J.; Barthel, H.; Akatsu, H.; Ouchi, Y.; Senda, K.; Murayama, S.; Ishii, K.; Takao, M.; et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: Phase 3 study. Alzheimer’s Dement. 2015, 11, 964–974. [Google Scholar] [CrossRef] [Green Version]
- Ikonomovic, M.D.; Buckley, C.J.; Heurling, K.; Sherwin, P.; Jones, P.; Zanette, M.; Mathis, C.A.; Klunk, W.E.; Chakrabarty, A.; Ironside, J.; et al. Post-mortem histopathology underlying β-amyloid PET imaging following flutemetamol F 18 injection. Acta Neuropathol. Commun. 2016, 4, 130. [Google Scholar] [CrossRef]
- Lemoine, L.; Gillberg, P.-G.; Bogdanovic, N.; Nennesmo, I.; Saint-Aubert, L.; Viitanen, M.; Graff, C.; Ingelsson, M.; Nordberg, A. Amyloid, tau, and astrocyte pathology in autosomal-dominant Alzheimer’s disease variants: AβPParc and PSEN1DE9. Mol. Psychiatry 2020, 26, 1–11. [Google Scholar] [CrossRef]
- Langheinrich, T.; Kobylecki, C.; Jones, M.; Thompson, J.C.; Snowden, J.S.; Hinz, R.; Pickering-Brown, S.; Mann, D.; Roncaroli, F.; Herholz, K.; et al. Amyloid-PET Positive Patient with bvFTD: Wrong Diagnosis, False Positive Scan, or Co-pathology? Neurol. Clin. Pr. 2021, 11, e952–e955. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, D.; Ossenkoppele, R., Jr.; Laforce, R. Evidence-based Interpretation of Amyloid-β PET Results. Alzheimer Dis. Assoc. Disord. 2018, 32, 28–34. [Google Scholar] [CrossRef]
- Spinelli, E.G.; Mandelli, M.L.; Miller, Z.A.; Santos-Santos, M.A.; Wilson, S.M.; Agosta, F.; Grinberg, L.T.; Huang, E.J.; Trojanowski, J.Q.; Meyer, M.; et al. Typical and atypical pathology in primary progressive aphasia variants. Ann. Neurol. 2017, 81, 430–443. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Jagust, W.J.; Furst, A.J.; Ms, J.M.O.; Racine, C.A.; Bs, E.C.M.; O’Neil, J.P.; Lal, R.A.; Dronkers, N.F.; Miller, B.L.; et al. A β amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann. Neurol. 2008, 64, 388–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, D.; Gorno-Tempini, M.L.; Rabinovici, G.D.; Santos-Santos, M.A.; Seeley, W.; Miller, B.L.; Pijnenburg, Y.; Keulen, M.A.; Groot, C.; van Berckel, B.N.M.; et al. Prevalence of amyloid-β pathology in distinct variants of primary progressive aphasia. Ann. Neurol. 2018, 84, 729–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Santos, M.A.; Rabinovici, G.D.; Iaccarino, L.; Ayakta, N.; Tammewar, G.; Lobach, I.; Henry, M.L.; Hubbard, I.; Mandelli, M.L.; Spinelli, E.G.; et al. Rates of Amyloid Imaging Positivity in Patients With Primary Progressive Aphasia. JAMA Neurol. 2018, 75, 342–352. [Google Scholar] [CrossRef] [Green Version]
- Villarejo-Galende, A.; Llamas-Velasco, S.; Gomez-Grande, A.; Puertas-Martin, V.; Contador, I.; Sarandeses, P.; González-Sánchez, M.; Trincado, R.; Pilkington, P.; Ruiz-Solis, S.; et al. Amyloid pet in primary progressive aphasia: Case series and systematic review of the literature. J. Neurol. 2016, 264, 121–130. [Google Scholar] [CrossRef]
- Jeon, S.; Kang, J.M.; Seo, S.; Jeong, H.J.; Funck, T.; Lee, S.-Y.; Park, K.H.; Lee, Y.-B.; Yeon, B.K.; Ido, T.; et al. Topographical Heterogeneity of Alzheimer’s Disease Based on MR Imaging, Tau PET, and Amyloid PET. Front. Aging Neurosci. 2019, 11, 211. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.D.; Josephs, K.A.; Machulda, M.M.; Drubach, D.A.; Apostolova, L.G.; Lowe, V.J.; Whitwell, J.L. Clinical, FDG and amyloid PET imaging in posterior cortical atrophy. J. Neurol. 2015, 262, 1483–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, L.C.; Corlier, F.; Habert, M.-O.; Uspenskaya, O.; Maroy, R.; Lamari, F.; Chupin, M.; Lehéricy, S.; Colliot, O.; Hahn-Barma, V.; et al. Similar amyloid-β burden in posterior cortical atrophy and Alzheimer’s disease. Brain 2011, 134, 2036–2043. [Google Scholar] [CrossRef]
- Martersteck, A.; Murphy, C.; Rademaker, A.; Wieneke, C.; Weintraub, S.; Chen, K.; Mesulam, M.-M.; Rogalski, E.; For the Alzheimer’s Disease Neuroimaging Initiative. Is in vivo amyloid distribution asymmetric in primary progressive aphasia? Ann. Neurol. 2016, 79, 496–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, M.; Ghosh, P.M.; Madison, C.; Laforce, R.J.; Corbetta-Rastelli, C.; Weiner, M.W.; Greicius, M.D.; Seeley, W.W.; Gorno-Tempini, M.L.; Rosen, H.J.; et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain 2013, 136, 844–858. [Google Scholar] [CrossRef]
- Vos, S.J.B.; Verhey, F.; Frölich, L.; Kornhuber, J.; Wiltfang, J.; Maier, W.; Peters, O.; Rüther, E.; Nobili, F.; Morbelli, S.; et al. Prevalence and prognosis of Alzheimer’s disease at the mild cognitive impairment stage. Brain 2015, 138, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Bilgel, M.; Beason-Held, L.; An, Y.; Zhou, Y.; Wong, D.F.; Resnick, S.M. Longitudinal evaluation of surrogates of regional cerebral blood flow computed from dynamic amyloid PET imaging. J. Cereb. Blood Flow Metab. 2019, 40, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Daerr, S.; Brendel, M.; Zach, C.; Mille, E.; Schilling, D.; Zacherl, M.J.; Bürger, K.; Danek, A.; Pogarell, O.; Schildan, A.; et al. Evaluation of early-phase [18F]-florbetaben PET acquisition in clinical routine cases. NeuroImage Clin. 2016, 14, 77–86. [Google Scholar] [CrossRef]
- Son, S.H.; Kang, K.; Ko, P.-W.; Lee, H.-W.; Lee, S.-W.; Ahn, B.-C.; Lee, J.; Yoon, U.; Jeong, S.Y. Early-Phase 18F-Florbetaben PET as an Alternative Modality for 18F-FDG PET. Clin. Nucl. Med. 2019, 45, e8–e14. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Yamaguchi, Y.; Mishra, S.K.; Higuchi, M.; Sahara, N. Tau Filaments and the Development of Positron Emission Tomography Tracers. Front. Neurol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleisher, A.S.; Pontecorvo, M.J.; Devous, M.D.; Lu, M.; Arora, A.K.; Truocchio, S.P.; Aldea, P.; Flitter, M.; Locascio, T.; Devine, M.; et al. Positron Emission Tomography Imaging With [18F]flortaucipir and Postmortem Assessment of Alzheimer Disease Neuropathologic Changes. JAMA Neurol. 2020, 77, 829. [Google Scholar] [CrossRef] [PubMed]
- Soleimani-Meigooni, D.N.; Iaccarino, L.; La Joie, R.; Baker, S.; Bourakova, V.; Boxer, A.L.; Edwards, L.; Eser, R.; Gorno-Tempini, M.-L.; Jagust, W.J.; et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain 2020, 143, 3477–3494. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Rabinovici, G.D.; Smith, R.; Cho, H.; Schöll, M.; Strandberg, O.; Palmqvist, S.; Mattsson-Carlgren, N.; Janelidze, S.; Santillo, A.; et al. Discriminative Accuracy of [18F]flortaucipir Positron Emission Tomography for Alzheimer Disease vs. Other Neurodegenerative Disorders. JAMA 2018, 320, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Hammes, J.; Bischof, G.N.; Bohn, K.P.; Onur, Ö.; Schneider, A.; Fliessbach, K.; Hoenig, M.C.; Jessen, F.; Neumaier, B.; Drzezga, A.E.; et al. One-Stop Shop: 18F-Flortaucipir PET Differentiates Amyloid-Positive and -Negative Forms of Neurodegenerative Diseases. J. Nucl. Med. 2020, 62, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Smith, R.; Ossenkoppele, R.; Santillo, A.; Borroni, E.; Klein, G.; Ohlsson, T.; Jögi, J.; Palmqvist, S.; Mattsson-Carlgren, N.; et al. Diagnostic Performance of RO948 F 18 Tau Positron Emission Tomography in the Differentiation of Alzheimer Disease From Other Neurodegenerative Disorders. JAMA Neurol. 2020, 77, 955. [Google Scholar] [CrossRef]
- Mueller, A.; Bullich, S.; Barret, O.; Madonia, J.; Berndt, M.; Papin, C.; Perrotin, A.; Koglin, N.; Kroth, H.; Pfeifer, A.; et al. Tau PET imaging with 18F-PI-2620 in Patients with Alzheimer Disease and Healthy Controls: A First-in-Humans Study. J. Nucl. Med. 2020, 61, 911–919. [Google Scholar] [CrossRef]
- Yap, S.Y.; Frias, B.; Wren, M.C.; Schöll, M.; Fox, N.C.; Årstad, E.; Lashley, T.; Sander, K. Discriminatory ability of next-generation tau PET tracers for Alzheimer’s disease. Brain 2021, 144, 2284–2290. [Google Scholar] [CrossRef]
- Beyer, L.; Brendel, M. Imaging of Tau Pathology in Neurodegenerative Diseases: An Update. Semin. Nucl. Med. 2021, 51, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Pascoal, T.A.; Strandberg, O.; Insel, P.; Smith, R.; Mattsson-Carlgren, N.; Benedet, A.L.; Cho, H.; Lyoo, C.H.; La Joie, R.; et al. A multicenter comparison of [18F]flortaucipir, [18F]RO948, and [18F]MK6240 tau PET tracers to detect a common target ROI for differential diagnosis. Eur. J. Pediatr. 2021, 48, 2295–2305. [Google Scholar] [CrossRef] [PubMed]
- Gogola, A.; Minhas, D.S.; Villemagne, V.L.; Cohen, A.D.; Mountz, J.M.; Pascoal, T.A.; Laymon, C.M.; Mason, N.S.; Ikonomovic, M.D.; Mathis, C.A.; et al. Direct Comparison of the Tau PET Tracers 18F-Flortaucipir and 18F-MK-6240 in Human Subjects. J. Nucl. Med. 2021, 63, 108–116. [Google Scholar] [CrossRef]
- Smith, R.; Schöll, M.; Leuzy, A.; Jögi, J.; Ohlsson, T.; Strandberg, O.; Hansson, O. Head-to-head comparison of tau positron emission tomography tracers [18F]flortaucipir and [18F]RO948. Eur. J. Pediatr. 2019, 47, 342–354. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.; Oh, S.J.; Lee, S.J.; Oh, J.S.; Roh, J.H.; Chung, S.J.; Lee, J.-H.; Lee, C.S.; Kim, J.S. Clinical Evaluation of 18F-PI-2620 as a Potent PET Radiotracer Imaging Tau Protein in Alzheimer Disease and Other Neurodegenerative Diseases Compared With 18F-THK-5351. Clin. Nucl. Med. 2020, 45, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Schonhaut, D.R.; Schöll, M.; Lockhart, S.N.; Ayakta, N.; Baker, S.L.; O’Neil, J.P.; Janabi, M.; Lazaris, A.; Cantwell, A.; et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016, 139, 1551–1567. [Google Scholar] [CrossRef] [Green Version]
- Tanner, J.A.; Rabinovici, G.D. Relationship Between Tau and Cognition in the Evolution of Alzheimer’s Disease: New Insights from Tau PET. J. Nucl. Med. 2020, 62, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Charil, A.; Shcherbinin, S.; Southekal, S.; Devous, M.D.; Mintun, M.; Murray, M.E.; Miller, B.B.; Schwarz, A.J. Tau Subtypes of Alzheimer’s Disease Determined in vivo Using Flortaucipir PET Imaging. J. Alzheimer’s Dis. 2019, 71, 1037–1048. [Google Scholar] [CrossRef]
- Schöll, M.; Ossenkoppele, R.; Strandberg, O.; Palmqvist, S.; Jögi, J.; Ohlsson, T.; Smith, R.; Hansson, O. The Swedish BioFINDER Study Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain 2017, 140, 2286–2294. [Google Scholar] [CrossRef]
- La Joie, R.; Visani, A.V.; Lesman-Segev, O.H.; Baker, S.L.; Edwards, L.; Iaccarino, L.; Soleimani-Meigooni, D.N.; Mellinger, T.; Janabi, M.; Miller, Z.A.; et al. Association of APOE4 and Clinical Variability in Alzheimer Disease With the Pattern of Tau- and Amyloid-PET. Neurology 2021, 96, e650–e661. [Google Scholar] [CrossRef]
- Sintini, I.; Martin, P.R.; Graff-Radford, J.; Senjem, M.L.; Schwarz, C.G.; Machulda, M.M.; Spychalla, A.J.; Drubach, D.A.; Knopman, D.S.; Petersen, R.C.; et al. Longitudinal tau-PET uptake and atrophy in atypical Alzheimer’s disease. NeuroImage Clin. 2019, 23, 101823. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Ba, D.R.S.; Baker, S.L.; O’Neil, J.P.; Janabi, M.; Ghosh, P.M.; Santos-Santos, M.A.; Miller, Z.; Bettcher, B.M.; Gorno-Tempini, M.L.; et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann. Neurol. 2014, 77, 338–342. [Google Scholar] [CrossRef]
- Sun, N.; Mormino, E.C.; Chen, J.; Sabuncu, M.R.; Yeo, B.T. Initiative ADN. Multi-modal latent factor exploration of atrophy, cognitive and tau heterogeneity in Alzheimer’s disease. NeuroImage 2019, 201, 116043. [Google Scholar] [CrossRef] [Green Version]
- Sintini, I.; Graff-Radford, J.; Senjem, M.L.; Schwarz, C.G.; Machulda, M.M.; Martin, P.R.; Jones, D.T.; Boeve, B.F.; Knopman, D.S.; Kantarci, K.; et al. Longitudinal neuroimaging biomarkers differ across Alzheimer’s disease phenotypes. Brain 2020, 143, 2281–2294. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Ms, P.R.M.; Botha, H.; Schwarz, C.G.; Duffy, J.R.; Clark, H.M.; Machulda, M.M.; Graff-Radford, J.; Ms, S.D.W.; Ms, M.L.S.; et al. [18F]AV-1451 tau-PET and primary progressive aphasia. Ann. Neurol. 2018, 83, 599–611. [Google Scholar] [CrossRef]
- Makaretz, S.J.; Quimby, M.; Collins, J.; Makris, N.; McGinnis, S.; Schultz, A.; Vasdev, N.; Johnson, K.A.; Dickerson, B.C. Flortaucipir tau PET imaging in semantic variant primary progressive aphasia. J. Neurol. Neurosurg. Psychiatry 2017, 89, 1024–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, R.M.; Bejanin, A.; Lesman-Segev, O.; Lajoie, R.; Visani, A.; Bourakova, V.; O’Neil, J.P.; Janabi, M.; Baker, S.; Lee, S.E.; et al. 18F-flortaucipir (AV-1451) tau PET in frontotemporal dementia syndromes. Alzheimer’s Res. Ther. 2019, 11, 1–18. [Google Scholar] [CrossRef]
- Whitwell, J.L.; Graff-Radford, J.; Tosakulwong, N.; Weigand, S.D.; Machulda, M.; Senjem, M.L.; Schwarz, C.J.; Spychalla, A.J.; Jones, D.T.; Drubach, D.A.; et al. [18F]AV-1451 clustering of entorhinal and cortical uptake in Alzheimer’s disease. Ann. Neurol. 2018, 83, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.W.; Initiative, T.A.D.N.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef]
- Aksman, L.M.; Oxtoby, N.P.; Scelsi, M.A.; Wijeratne, P.A.; Young, A.L.; Lopes Alves, I.; Barkhof, F.; Alexander, D.C.; Altmann, A. Tau-First Subtype of Alzheimer’s Disease Consistently Identified across In Vivo and Post Mortem Studies [Internet]. Neuroscience. December 2020. Available online: http://biorxiv.org/lookup/doi/10.1101/2020.12.18.418004 (accessed on 12 November 2021).
- Schöll, M.; Lockhart, S.N.; Schonhaut, D.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.; et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 2016, 89, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, J.W.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; Alzheimer’s Disease Neuroimaging Initiative. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef] [PubMed]
- Franzmeier, N.; Dewenter, A.; Frontzkowski, L.; Dichgans, M.; Rubinski, A.; Neitzel, J.; Smith, R.; Strandberg, O.; Ossenkoppele, R.; Buerger, K.; et al. Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer’s disease. Sci. Adv. 2020, 6, eabd1327. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Kim, Y.J.; Lee, H.S.; Lee, J.H.; Ryu, Y.H.; Lee, M.S.; Lyoo, C.H. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann. Neurol. 2016, 80, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Cope, T.E.; Rittman, T.; Borchert, R.J.; Jones, P.S.; Vatansever, D.; Allinson, K.; Passamonti, L.; Vazquez Rodriguez, P.; Bevan-Jones, W.R.; O’Brien, J.T.; et al. Tau burden and the functional connectome in Alzheimer’s disease and progressive supranuclear palsy. Brain 2018, 141, 550–567. [Google Scholar] [CrossRef]
- Pascual, B.; Funk, Q.; Zanotti-Fregonara, P.; Pal, N.; Rockers, E.; Yu, M.; Spann, B.; Román, G.C.; Schulz, P.E.; Karmonik, C.; et al. Multimodal 18F-AV-1451 and MRI Findings in Nonfluent Variant of Primary Progressive Aphasia: Possible Insights on Nodal Propagation of Tau Protein Across the Syntactic Network. J. Nucl. Med. 2019, 61, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Seemiller, J.; Bischof, G.N.; Hoenig, M.C.; Tahmasian, M.; van Eimeren, T.; Drzezga, A.; Initiative, A.T.A.D.N. Indication of retrograde tau spreading along Braak stages and functional connectivity pathways. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- Ballarini, T.; Iaccarino, L.; Magnani, G.; Ayakta, N.; Miller, B.L.; Jagust, W.J.; Gorno-Tempini, M.L.; Rabinovici, G.D.; Perani, D. Neuropsychiatric subsyndromes and brain metabolic network dysfunctions in early onset Alzheimer’s disease. Hum. Brain Mapp. 2016, 37, 4234–4247. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, F.; Orini, S.; Gandolfo, F.; Altomare, D.; Festari, C.; Agosta, F.; Arbizu, J.; Drzezga, A.; Nestor, P.; Nobili, F.; et al. Diagnostic utility of FDG-PET in the differential diagnosis between different forms of primary progressive aphasia. Eur. J. Pediatr. 2018, 45, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Kalpouzos, G.; Eustache, F.; De La Sayette, V.; Viader, F.; Chételat, G.; Desgranges, B. Working memory and FDG–PET dissociate early and late onset Alzheimer disease patients. J. Neurol. 2005, 252, 548–558. [Google Scholar] [CrossRef]
- Mosconi, L.; Tsui, W.-H.; De Santi, S.; Li, J.; Rusinek, H.; Convit, A.; Li, Y.; Boppana, M.; de Leon, M.J. Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology 2005, 64, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Villain, N.; Fouquet, M.; Baron, J.-C.; Mézenge, F.; Landeau, B.; de La Sayette, V.; Viader, F.; Eustache, F.; Desgranges, B.; Chételat, G. Sequential relationships between grey matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer’s disease. Brain 2010, 133, 3301–3314. [Google Scholar] [CrossRef] [Green Version]
- Nestor, P.; Caine, D.; Fryer, T.D.; Clarke, J.; Hodges, J.R. The topography of metabolic deficits in posterior cortical atrophy (the visual variant of Alzheimer’s disease) with FDG-PET. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1521–1529. [Google Scholar] [CrossRef]
- Graff-Radford, J.; Murray, M.E.; Lowe, V.J.; Boeve, B.F.; Ferman, T.J.; Przybelski, S.A.; Lesnick, T.G.; Senjem, M.L.; Gunter, J.L.; Smith, G.E.; et al. Dementia with Lewy bodies: Basis of cingulate island sign. Neurology 2014, 83, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Whitwell, J.L.; Graff-Radford, J.; Singh, T.D.; Drubach, D.A.; Senjem, M.L.; Spychalla, A.J.; Tosakulwong, N.; Lowe, V.J.; Josephs, K.A. 18F-FDG PET in Posterior Cortical Atrophy and Dementia with Lewy Bodies. J. Nucl. Med. 2016, 58, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Verma, R.; Ranjan, R.; Belho, E.S.; Seniaray, N.; Dinand, V.; Malik, D.; Mahajan, H. Metabolic imaging patterns in posterior cortical atrophy and Lewy body dementia. Nucl. Med. Commun. 2019, 40, 1275–1282. [Google Scholar] [CrossRef]
- Madhavan, A.; Whitwell, J.L.; Weigand, S.D.; Duffy, J.R.; Strand, E.A.; Machulda, M.M.; Tosakulwong, N.; Senjem, M.; Gunter, J.L.; Lowe, V.J.; et al. FDG PET and MRI in Logopenic Primary Progressive Aphasia versus Dementia of the Alzheimer’s Type. PLoS ONE 2013, 8, e62471. [Google Scholar] [CrossRef]
- Matías-Guiu, J.A.; Cabrera-Martín, M.N.; Moreno-Ramos, T.; Valles-Salgado, M.; Fernandez-Matarrubia, M.; Carreras, J.L.; Matías-Guiu, J. Amyloid and FDG-PET study of logopenic primary progressive aphasia: Evidence for the existence of two subtypes. J. Neurol. 2015, 262, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Duffy, J.R.; Strand, E.A.; Machulda, M.M.; Vemuri, P.; Senjem, M.L.; Nobili, R.P.; Baker, M.C.; Lowe, V.; Jack, C.R., Jr.; et al. Progranulin-associated PiB-negative logopenic primary progressive aphasia. J Neurol. 2014, 261, 604–614. [Google Scholar] [CrossRef] [Green Version]
- Woodward, M.C.; Rowe, C.C.; Jones, G.; Villemagne, V.L.; Varos, T.A. Differentiating the Frontal Presentation of Alzheimer’s Disease with FDG-PET. J. Alzheimer’s Dis. 2015, 44, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Singleton, E.H.; Pijnenburg, Y.A.; Sudre, C.H.; Groot, C.; Kochova, E.; Barkhof, F.; Joie, R.L.; Rosen, H.J.; Seeley, W.W.; Miller, B.; et al. Investigating the clinico-anatomical dissociation in the behavioral variant of Alzheimer disease. Alzheimers Res. Ther. 2020, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Townley, R.A.; Graff-Radford, J.; Mantyh, W.G.; Botha, H.; Polsinelli, A.J.; Przybelski, S.A.; Machulda, M.M.; Makhlouf, A.T.; Senjem, M.L.; Murray, M.E.; et al. Progressive dysexecutive syndrome due to Alzheimer’s disease: A description of 55 cases and comparison to other phenotypes. Brain Commun. 2020, 2, fcaa068. [Google Scholar] [CrossRef] [PubMed]
- Herholz, K. PET studies in dementia. Ann. Nucl. Med. 2003, 17, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Scialò, C.; Ferrara, M.; Accardo, J.; Morbelli, S.; Picco, A.; Arnaldi, D.; Brugnolo, A.; Girtler, N.; Nobili, F. Frontal variant Alzheimer disease or frontotemporal lobe degeneration with incidental amyloidosis? Alzheimer Dis. Assoc. Disord. 2016, 30, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Levin, F.; Ferreira, D.; Lange, C.; Dyrba, M.; Westman, E.; Buchert, R.; Teipel, S.J.; Grothe, M.J.; Initiative, F.T.A.D.N. Data-driven FDG-PET subtypes of Alzheimer’s disease-related neurodegeneration. Alzheimer’s Res. Ther. 2021, 13, 49. [Google Scholar] [CrossRef]
- Yakushev, I.; Drzezga, A.; Habeck, C. Metabolic connectivity: Methods and applications. Curr. Opin. Neurol. 2017, 30, 677–685. [Google Scholar] [CrossRef]
- Herholz, K.; Haense, C.; Gerhard, A.; Jones, M.; Anton-Rodriguez, J.; Segobin, S.; Snowden, J.S.; Thompson, J.C.; Kobylecki, C. Metabolic regional and network changes in Alzheimer’s disease subtypes. J. Cereb. Blood Flow Metab. 2018, 38, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Dolui, S.; Li, Z.; Nasrallah, I.M.; Detre, J.A.; Wolk, D.A. Arterial spin labeling versus 18F-FDG-PET to identify mild cognitive impairment. NeuroImage Clin. 2019, 25, 102146. [Google Scholar] [CrossRef]
- Wong, C.-Y.O.; Thie, J.; Gaskill, M.; Ponto, R.; Hill, J.; Tian, H.-Y.; Balon, H.; Wu, D.; Fink-Bennett, D.; Nagle, C. A statistical investigation of normal regional intra-subject heterogeneity of brain metabolism and perfusion by F-18 FDG and O-15 H2O PET imaging. BMC Nucl. Med. 2006, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannsen, P.; Jakobsen, J.; Gjedde, A. Statistical maps of cerebral blood flow deficits in Alzheimer’s disease. Eur. J. Neurol. 2000, 7, 385–392. [Google Scholar] [CrossRef]
- Waldemar, G.; Bruhn, P.; Kristensen, M.; Johnsen, A.; Paulson, O.B.; Lassen, N.A. Heterogeneity of neocortical cerebral blood flow deficits in dementia of the Alzheimer type: A [99mTc]-d,l-HMPAO SPECT study. J. Neurol. Neurosurg. Psychiatry 1994, 57, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wolk, D.A.; Reddin, J.S.; Korczykowski, M.; Martinez, P.M.; Musiek, E.S.; Newberg, A.B.; Julin, P.; Arnold, S.E.; Greenberg, J.H.; et al. Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology 2011, 77, 1977–1985. [Google Scholar] [CrossRef] [Green Version]
- Verclytte, S.; Lopes, R.; Lenfant, P.; Rollin, A.; Semah, F.; Leclerc, X.; Pasquier, F.; Delmaire, C. Cerebral Hypoperfusion and Hypometabolism Detected by Arterial Spin Labeling MRI and FDG-PET in Early-Onset Alzheimer’s Disease. J. Neuroimaging 2015, 26, 207–212. [Google Scholar] [CrossRef]
- Cha, Y.-H.K.; Jog, M.A.; Kim, Y.-C.; Chakrapani, S.; Kraman, S.M.; Wang, D. Regional Correlation between Resting State FDG PET and pCASL Perfusion MRI. Br. J. Pharmacol. 2013, 33, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.T.; Firbank, M.J.; Davison, C.; Barnett, N.; Bamford, C.; Donaldson, C.; Olsen, K.; Herholz, K.; Williams, D.; Lloyd, J. 18F-FDG PET and Perfusion SPECT in the Diagnosis of Alzheimer and Lewy Body Dementias. J. Nucl. Med. 2014, 55, 1959–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riederer, I.; Bohn, K.P.; Preibisch, C.; Wiedemann, E.; Zimmer, C.; Alexopoulos, P.; Förster, S. Alzheimer Disease and Mild Cognitive Impairment: Integrated Pulsed Arterial Spin-Labeling MRI and 18F-FDG PET. Radiology 2018, 288, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fällmar, D.; Haller, S.; Lilja, J.; Danfors, T.; Kilander, L.; Tolboom, N.; Egger, K.; Kellner, E.; Croon, P.M.; Verfaillie, S.; et al. Arterial spin labeling-based Z-maps have high specificity and positive predictive value for neurodegenerative dementia compared to FDG-PET. Eur. Radiol. 2017, 27, 4237–4246. [Google Scholar] [CrossRef]
- Seeley, W.W.; Crawford, R.K.; Zhou, J.; Miller, B.L.; Greicius, M.D. Neurodegenerative Diseases Target Large-Scale Human Brain Networks. Neuron 2009, 62, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-Y.; Initiative, A.D.N.; Na, H.K.; Kim, S.; Kim, H.; Kim, H.J.; Seo, S.W.; Na, D.L.; Han, C.E.; Seong, J.-K.; et al. Robust Identification of Alzheimer’s Disease subtypes based on cortical atrophy patterns. Sci. Rep. 2017, 7, 43270. [Google Scholar] [CrossRef] [Green Version]
- Chételat, G.; Landeau, B.; Salmon, E.; Yakushev, I.; Bahri, M.A.; Mézenge, F.; Perrotin, A.; Bastin, C.; Manrique, A.; Scheurich, A.; et al. Relationships between brain metabolism decrease in normal aging and changes in structural and functional connectivity. NeuroImage 2013, 76, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risacher, S.L.; Anderson, W.H.; Charil, A.; Castelluccio, P.F.; Shcherbinin, S.; Saykin, A.J.; Schwarz, A.J.; Initiative, F.T.A.D.N. Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology 2017, 89, 2176–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, H.K.; Kang, D.R.; Kim, S.; Seo, S.W.; Heilman, K.M.; Noh, Y.; Na, D.L. Malignant progression in parietal-dominant atrophy subtype of Alzheimer’s disease occurs independent of onset age. Neurobiol. Aging 2016, 47, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Planche, V.; Coupé, P.; Helmer, C.; Le Goff, M.; Amieva, H.; Tison, F.; Dartigues, J.-F.; Catheline, G. Evolution of brain atrophy subtypes during aging predicts long-term cognitive decline and future Alzheimer’s clinical syndrome. Neurobiol. Aging 2019, 79, 22–29. [Google Scholar] [CrossRef]
- Zhang, B.; Lin, L.; Wu, S. A Review of Brain Atrophy Subtypes Definition and Analysis for Alzheimer’s Disease Heterogeneity Studies. J. Alzheimer’s Dis. 2021, 80, 1339–1352. [Google Scholar] [CrossRef]
- Dong, A.; Toledo, J.B.; Honnorat, N.; Doshi, J.; Varol, E.; Sotiras, A.; Wolk, D.; Trojanowski, J.Q.; Davatzikos, C. Heterogeneity of neuroanatomical patterns in prodromal Alzheimer’s disease: Links to cognition, progression and biomarkers. Brain 2017, 140, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Park, J.-Y.; Seo, S.W.; Jung, Y.H.; Kim, Y.; Jang, H.; Kim, S.T.; Seong, J.-K.; Na, D.L. Cortical atrophy pattern–based subtyping predicts prognosis of amnestic MCI: An individual-level analysis. Neurobiol. Aging 2018, 74, 38–45. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Lyoo, C.H.; Sudre, C.H.; van Westen, D.; Cho, H.; Ryu, Y.H.; Choi, J.Y.; Smith, R.; Strandberg, O.; Palmqvist, S.; et al. Distinct tau PET patterns in atrophy-defined subtypes of Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 335–344. [Google Scholar] [CrossRef]
- Das, S.R.; Lyu, X.; Duong, M.T.; Xie, L.; McCollum, L.; de Flores, R.; DiCalogero, M.; Irwin, D.J.; Dickerson, B.C.; Nasrallah, I.M.; et al. Tau-Atrophy Variability Reveals Phenotypic Heterogeneity in Alzheimer’s Disease. Ann. Neurol. 2021, 90, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Marinescu, R.V.; Oxtoby, N.P.; Bocchetta, M.; Yong, K.; Firth, N.C.; Cash, D.M.; Thomas, D.L.; Dick, K.M.; Cardoso, J.; et al. Uncovering the heterogeneity and temporal complexity of neurodegenerative diseases with Subtype and Stage Inference. Nat. Commun. 2018, 15, 4273. [Google Scholar] [CrossRef] [Green Version]
- Archetti, D.; Young, A.L.; Oxtoby, N.P.; Ferreira, D.; Mårtensson, G.; Westman, E.; Alexander, D.C.; Frisoni, G.B.; Redolfi, A. For Alzheimer’s Disease Neuroimaging Initiative and EuroPOND Consortium Inter-Cohort Validation of SuStaIn Model for Alzheimer’s Disease. Front. Big Data 2021, 4, 661110. [Google Scholar] [CrossRef]
- Marinescu, R.V.; Eshaghi, A.; Lorenzi, M.; Young, A.L.; Oxtoby, N.P.; Garbarino, S.; Crutch, S.J.; Alexander, D.C. DIVE: A spatiotemporal progression model of brain pathology in neurodegenerative disorders. NeuroImage 2019, 192, 166–177. [Google Scholar] [CrossRef]
- Marinescu, R.V.; Oxtoby, N.P.; Young, A.L.; Bron, E.E.; Toga, A.W.; Weiner, M.W.; Barkhof, F.; Fox, N.C.; Eshaghi, A.; Toni, T.; et al. The Alzheimer’s Disease Prediction Of Longitudinal Evolution (TADPOLE) Challenge: Results after 1 Year Follow-up. arXiv 2020, arXiv:2002.03419. Available online: http://arxiv.org/abs/2002.03419 (accessed on 10 February 2021).
- Giorgio, J.; Landau, S.M.; Jagust, W.J.; Tino, P.; Kourtzi, Z. Modelling prognostic trajectories of cognitive decline due to Alzheimer’s disease. NeuroImage Clin. 2020, 26, 102199. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Basaia, S.; Canu, E.; Imperiale, F.; Magnani, G.; Falautano, M.; Comi, G.; Falini, A.; Agosta, F. Changes in functional and structural brain connectome along the Alzheimer’s disease continuum. Mol. Psychiatry 2018, 25, 230–239. [Google Scholar] [CrossRef]
- Jacobs, H.I.L.; Hedden, T.; Schultz, A.P.; Sepulcre, J.; Perea, R.D.; Amariglio, R.E.; Papp, K.V.; Rentz, D.M.; Sperling, R.A.; Johnson, K.A. Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat. Neurosci. 2018, 21, 424–431. [Google Scholar] [CrossRef] [PubMed]
- King-Robson, J.; Wilson, H.; Politis, M. Alzheimer’s Disease Neuroimaging Initiative. Associations Between Amyloid and Tau Pathology, and Connectome Alterations, in Alzheimer’s Disease and Mild Cognitive Impairment. J. Alzheimer’s Dis. JAD 2021, 82, 541–560. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Small vessel disease: Mechanisms and clinical implications. Lancet Neurol. 2019, 18, 684–696. [Google Scholar] [CrossRef]
- Heiss, W.-D.; Rosenberg, G.A.; Thiel, A.; Berlot, R.; De Reuck, J. Neuroimaging in vascular cognitive impairment: A state-of-the-art review. BMC Med. 2016, 14, 170. [Google Scholar] [CrossRef] [Green Version]
- Razek, A.A.K.A.; Elsebaie, N.A. Imaging of vascular cognitive impairment. Clin. Imaging 2021, 74, 45–54. [Google Scholar] [CrossRef]
- Bonilha, L.; Gleichgerrcht, E.; Fridriksson, J.; Rorden, C.; Breedlove, J.L.; Nesland, T.; Paulus, W.; Helms, G.; Focke, N.K. Reproducibility of the Structural Brain Connectome Derived from Diffusion Tensor Imaging. PLoS ONE 2015, 10, e0135247. [Google Scholar] [CrossRef] [PubMed]
- Cerami, C.; Crespi, C.; Della Rosa, P.A.; Dodich, A.; Marcone, A.; Magnani, G.; Coppi, E.; Falini, A.; Cappa, S.F.; Perani, D. Brain Changes within the Visuo-Spatial Attentional Network in Posterior Cortical Atrophy. J. Alzheimer’s Dis. 2014, 43, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, N.C.; Primativo, S.; Marinescu, R.-V.; Shakespeare, T.J.; Suarez-Gonzalez, A.; Lehmann, M.; Carton, A.; Ocal, D.; Pavisic, I.; Paterson, R.W.; et al. Longitudinal neuroanatomical and cognitive progression of posterior cortical atrophy. Brain 2019, 142, 2082–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyton, C.E.; Hodges, J.R. Towards a Clearer Definition of Logopenic Progressive Aphasia. Curr. Neurol. Neurosci. Rep. 2013, 13, 396. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, J.D.; Ridgway, G.; Crutch, S.; Hailstone, J.; Goll, J.C.; Clarkson, M.; Mead, S.; Beck, J.; Mummery, C.; Ourselin, S.; et al. Progressive logopenic/phonological aphasia: Erosion of the language network. NeuroImage 2010, 49, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greicius, M.D.; Supekar, K.; Menon, V.; Dougherty, R.F. Resting-State Functional Connectivity Reflects Structural Connectivity in the Default Mode Network. Cereb. Cortex 2008, 19, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.W.; Guidon, A.; Doraiswamy, P.M.; Choudhury, K.R.; Liu, C.; Petrella, J.R.; Initiative, A.D.N.; Initiative, F.T.A.D.N. The Alzheimer Structural Connectome: Changes in Cortical Network Topology with Increased Amyloid Plaque Burden. Radiology 2014, 273, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Pasquini, L.; Benson, G.; Grothe, M.J.; Utz, L.; Myers, N.E.; Yakushev, I.; Grimmer, T.; Scherr, M.; Sorg, C. Individual Correspondence of Amyloid-β and Intrinsic Connectivity in the Posterior Default Mode Network Across Stages of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 763–773. [Google Scholar] [CrossRef]
- Jones, D.T.; Knopman, D.S.; Gunter, J.L.; Graff-Radford, J.; Vemuri, P.; Boeve, B.F.; Petersen, R.C.; Weiner, M.W.; Jack, C.R. Cascading network failure across the Alzheimer’s disease spectrum. Brain 2015, 139, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Scherr, M.; Utz, L.; Tahmasian, M.; Pasquini, L.; Grothe, M.J.; Rauschecker, J.P.; Grimmer, T.; Drzezga, A.; Sorg, C.; Riedl, V. Effective connectivity in the default mode network is distinctively disrupted in Alzheimer’s disease—A simultaneous resting-state FDG-PET/fMRI study. Hum. Brain Mapp. 2021, 42, 4134–4143. [Google Scholar] [CrossRef]
- van den Heuvel, M.P.; Sporns, O. A cross-disorder connectome landscape of brain dysconnectivity. Nat. Rev. Neurosci. 2019, 20, 435–446. [Google Scholar] [CrossRef]
- Bozzali, M.; Dowling, C.; Serra, L.; Spanò, B.; Torso, M.; Marra, C.; Castelli, D.; Dowell, N.G.; Koch, G.; Caltagirone, C.; et al. The Impact of Cognitive Reserve on Brain Functional Connectivity in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 44, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Grothe, M.; Strandberg, T.O.; Ohlsson, T.; Hägerström, D.; Jögi, J.; Smith, R.; Schöll, M. Tau Pathology Distribution in Alzheimer’s disease Corresponds Differentially to Cognition-Relevant Functional Brain Networks. Front. Neurosci. 2017, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M.C.; Bischof, G.N.; Seemiller, J.; Hammes, J.; Kukolja, J.; Onur, O.; Jessen, F.; Fliessbach, K.; Neumaier, B.; Fink, G.R.; et al. Networks of tau distribution in Alzheimer’s disease. Brain 2018, 141, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Veldsman, M.; Zamboni, G.; Butler, C.; Ahmed, S. Attention network dysfunction underlies memory impairment in posterior cortical atrophy. NeuroImage Clin. 2019, 22, 101773. [Google Scholar] [CrossRef] [PubMed]
- Glick-Shames, H.; Backner, Y.; Bick, A.; Raz, N.; Levin, N. The impact of localized grey matter damage on neighboring connectivity: Posterior cortical atrophy and the visual network. Brain Imaging Behav. 2018, 13, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Mandic-Stojmenovic, G.; Canu, E.; Stojkovic, T.; Imperiale, F.; Caso, F.; Stefanova, E.; Copetti, M.; Kostic, V.S.; Filippi, M. Functional and structural brain networks in posterior cortical atrophy: A two-centre multiparametric MRI study. NeuroImage Clin. 2018, 19, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, R.; Gallea, C.; Kas, A.; Perlbarg, V.; Samri, D.; Trotta, L.; Michon, A.; Lacomblez, L.; Dubois, B.; Lehericy, S.; et al. Functional Connectivity of Ventral and Dorsal Visual Streams in Posterior Cortical Atrophy. J. Alzheimer’s Dis. 2016, 51, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitwell, J.L.; Jones, D.T.; Duffy, J.R.; Strand, E.A.; Machulda, M.M.; Przybelski, S.A.; Vemuri, P.; Gregg, B.E.; Gunter, G.L.; Senjem, M.L.; et al. Working memory and language network dysfunctions in logopenic aphasia: A task-free fMRI comparison with Alzheimer’s dementia. Neurobiol. Aging. 2015, 36, 1245–1252. [Google Scholar] [CrossRef] [Green Version]
- Bonakdarpour, B.; Hurley, R.S.; Wang, A.R.; Fereira, H.R.; Basu, A.; Chatrathi, A.; Guillaume, K.; Rogalski, E.J.; Mesulam, M.M. Perturbations of language network connectivity in primary progressive aphasia. Cortex 2019, 121, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Ficek, B.; Rapp, B.; Tsapkini, K. Different patterns of functional network reorganization across the variants of primary progressive aphasia: A graph-theoretic analysis. Neurobiol. Aging 2020, 96, 184–196. [Google Scholar] [CrossRef]
- Drzezga, A. The Network Degeneration Hypothesis: Spread of Neurodegenerative Patterns Along Neuronal Brain Networks. J. Nucl. Med. 2018, 59, 1645–1648. [Google Scholar] [CrossRef]
- Hu, X.S.; Okamura, N.; Arai, H.; Higuchi, M.; Matsui, T.; Tashiro, M.; Shinkawa, M.; Itoh, M.; Ido, T.; Sasaki, H. 18F-fluorodopa PET study of striatal dopamine uptake in the diagnosis of dementia with Lewy bodies. Neurology 2000, 55, 1575–1577. [Google Scholar] [CrossRef]
- McKeith, I.; O’Brien, J.; Walker, Z.; Tatsch, K.; Booij, J.; Darcourt, J.; Padovani, A.; Giubbini, R.; Bonuccelli, U.; Volterrani, D.; et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: A phase III, multicentre study. Lancet Neurol. 2007, 6, 305–313. [Google Scholar] [CrossRef]
- Kadir, A.; Nordberg, A. Target-Specific PET Probes for Neurodegenerative Disorders Related to Dementia. J. Nucl. Med. 2010, 51, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Klein, J.C.; Eggers, C.; Kalbe, E.; Weisenbach, S.; Hohmann, C.; Vollmar, S.; Baudrexel, S.; Diederich, N.J.; Heiss, W.D.; Hilker, R. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology 2010, 74, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Shinotoh, H.; Shimada, H.; Ota, T.; Sato, K.; Tanaka, N.; Zhang, M.-R.; Higuchi, M.; Fukushi, K.; Irie, T.; et al. Voxel-Based Acetylcholinesterase PET Study in Early and Late Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.C.; Li, C.-S.R. Noradrenergic Dysfunction in Alzheimer’s and Parkinson’s Diseases—An Overview of Imaging Studies. Front. Aging Neurosci. 2018, 10, 127. [Google Scholar] [CrossRef]
- Terada, T.; Therriault, J.; Kang, M.S.P.; Savard, M.; Pascoal, T.A.; Lussier, F.; Tissot, C.; Wang, Y.-T.; Benedet, A.; Matsudaira, T.; et al. Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 1–12. [Google Scholar] [CrossRef]
- Mecca, A.P.; Chen, M.; O’Dell, R.S.; Naganawa, M.; Toyonaga, T.; Godek, T.A.; Harris, J.E.; Bartlett, H.H.; Zhao, W.; Nabulsi, N.B.; et al. In vivo measurement of widespread synaptic loss in Alzheimer’s disease with SV2A PET. Alzheimer’s Dement. 2020, 16, 974–982. [Google Scholar] [CrossRef]
- Bellaver, B.; Ferrari-Souza, J.P.; da Ros, L.U.; Carter, S.F.; Rodriguez-Vieitez, E.; Nordberg, A.; Pellerin, L.; Rosa-Neto, P.; Leffa, D.T.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer Disease. Neurology 2021, 96, e2944–e2955. [Google Scholar] [CrossRef]
- Dani, M.; Wood, M.; Mizoguchi, R.; Fan, Z.; Walker, Z.; Morgan, R.; Hinz, R.; Biju, M.; Kuruvilla, T.; Brooks, D.; et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 2018, 141, 2740–2754. [Google Scholar] [CrossRef]

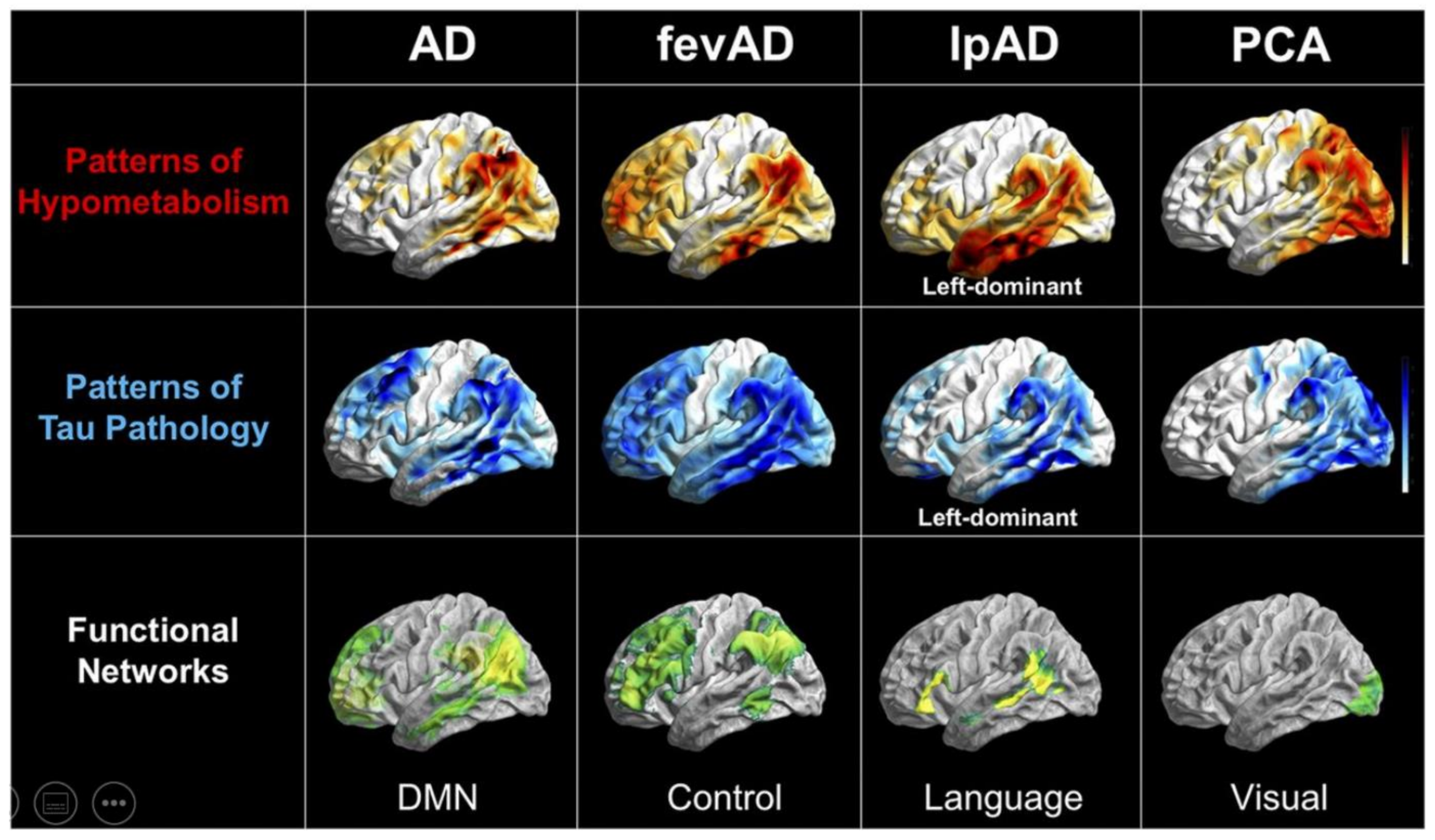
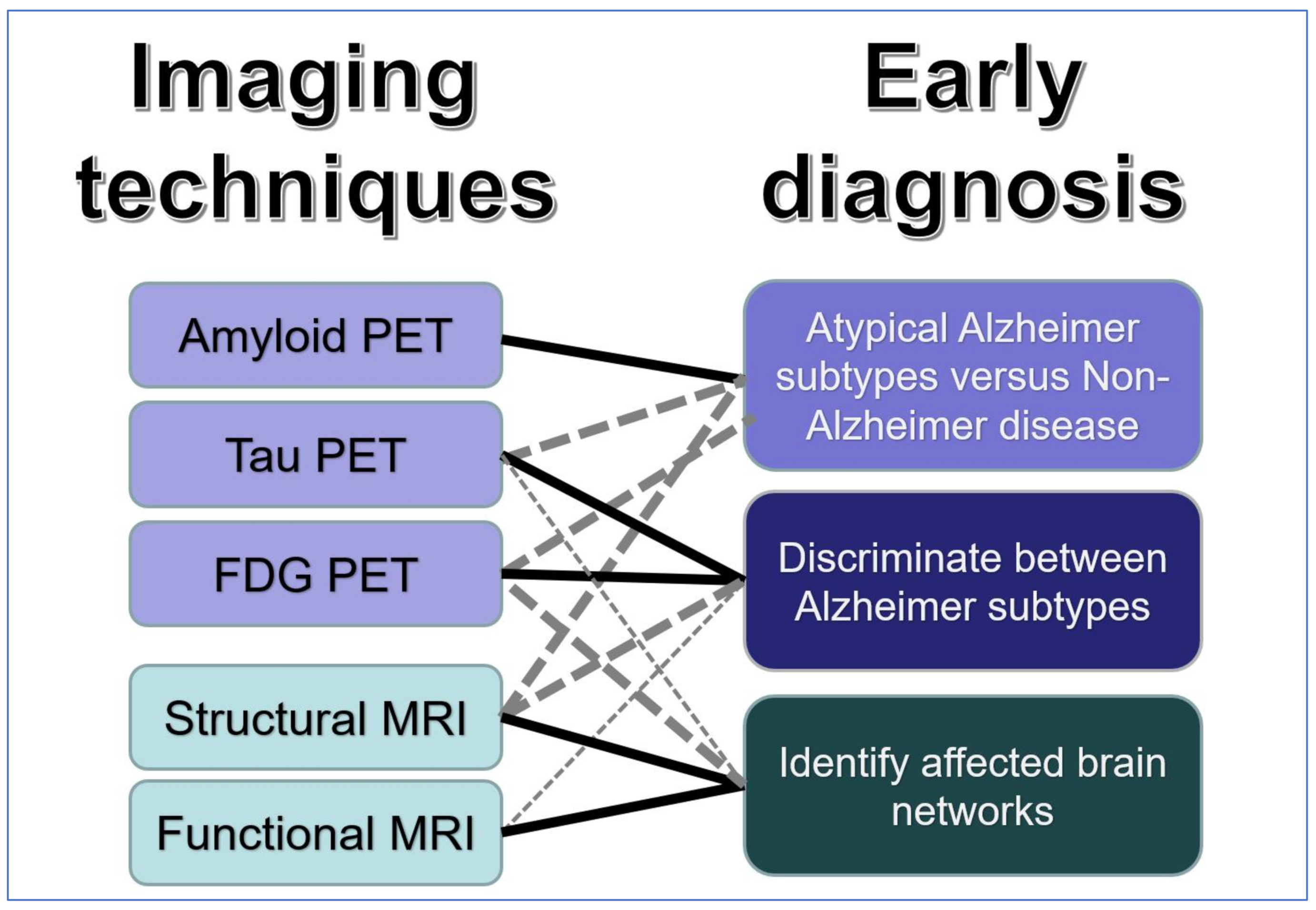
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herholz, K. Imaging Clinical Subtypes and Associated Brain Networks in Alzheimer’s Disease. Brain Sci. 2022, 12, 146. https://doi.org/10.3390/brainsci12020146
Herholz K. Imaging Clinical Subtypes and Associated Brain Networks in Alzheimer’s Disease. Brain Sciences. 2022; 12(2):146. https://doi.org/10.3390/brainsci12020146
Chicago/Turabian StyleHerholz, Karl. 2022. "Imaging Clinical Subtypes and Associated Brain Networks in Alzheimer’s Disease" Brain Sciences 12, no. 2: 146. https://doi.org/10.3390/brainsci12020146
APA StyleHerholz, K. (2022). Imaging Clinical Subtypes and Associated Brain Networks in Alzheimer’s Disease. Brain Sciences, 12(2), 146. https://doi.org/10.3390/brainsci12020146