
Hyperammonaemic Encephalopathy Caused by Adult-Onset Ornithine Transcarbamylase Deficiency

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
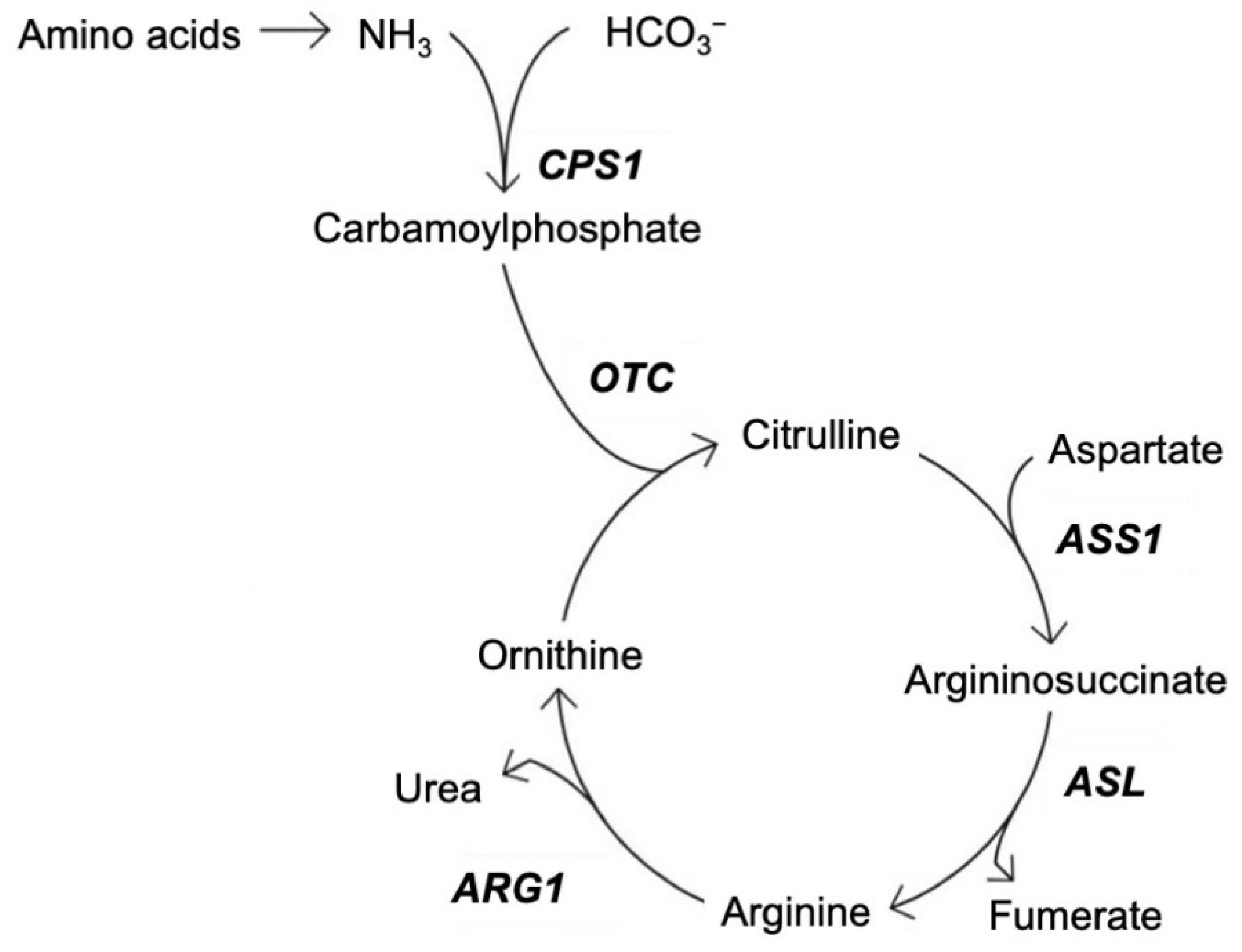
:1. Introduction
- (I)
- An overproduction of ammonia due to: (i) excessive protein load originating from conditions such as gastrointestinal haemorrhage, gastric bypass surgery, multiple myeloma, allogenic stem cell transplantation, or parenteral nutrition; (ii) an increased metabolism caused by starvation, seizures, vigorous exercise, burn injuries, or corticosteroid treatment; or (iii) urinary overproduction caused by urease-producing infections [1,2,3].
- (II)
- A reduced elimination of ammonia due to (i) liver failure caused by hepatitis or alcohol abuse; (ii) drugs such as antiepileptic drugs [6,7,8,9,10,11], pain medication [11,12], acetazolamide [13], chemotherapy [14], and methamphetamine [15]; or (iii) inborn errors of metabolism (urea cycle disorders (UCDs), organic acidaemias or fatty acid oxidation disorders) [16].
2. Case Report
3. Genetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaffalitzky De Muckadell, O.B.; Haunsø, S.; Vilstrup, S. Medicinsk Kompendium, 18; Nyt Nordisk Forlag Arnold Busck: København, Norway, 2013. [Google Scholar]
- Walker, V. Severe hyperammonaemia in adults not explained by liver disease. Ann. Clin. Biochem. 2012, 49, 214–228. [Google Scholar] [CrossRef]
- Sakusic, A.; Sabov, M.; McCambridge, A.J.; Rabinstein, A.A.; Singh, T.D.; Mukesh, K.; Kashani, K.B.; Cook, D.; Gajic, O. Features of Adult Hyperammonemia Not Due to Liver Failure in the I.C.U. Crit. Care Med. 2018, 46, e897–e903. [Google Scholar] [CrossRef] [PubMed]
- Walker, V. Ammonia metabolism and hyperammonemic disorders. Adv. Clin. Chem. 2014, 67, 73–150. [Google Scholar] [PubMed]
- Stergachis, A.B.; Mogensen, K.M.; Khoury, C.C.; Lin, A.P.; Peake, R.W.; Baker, J.J.; Barkoudah, E.; Sahai, I.; Sweetser, D.A.; Berry, G.T.; et al. A retrospective study of adult patients with noncirrhotic hyperammonemia. J. Inherit. Metab. Dis. 2020, 43, 1165–1172. [Google Scholar] [CrossRef]
- Aires, C.C.; van Cruchten, A.; Ijlst, L.; de Almeida, I.T.; Duran, M.; Wanders, R.J.; Silva, M.F.D. New insights on the mechanisms of valproate-induced hyperammonemia: Inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J. Hepatol. 2011, 55, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Ambrosetto, G.; Riva, R.; Baruzzi, A. Hyperammonemia in asterixis induced by carbamazepine: Two case reports. Acta Neurol. Scand. 1984, 69, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ibañez, A.; Urrestarazu-Bolumburu, E.; Viteri-Torres, C. Hyperammonemic encephalopathy related to valproate, phenobarbital, and topiramate synergism. Epilepsy Behav. 2011, 21, 480–482. [Google Scholar] [CrossRef]
- Hansen, N.; Finzel, M.; Block, F. [Antiepileptic drug-induced encephalopathy]. Fortschr. Neurol. Psychiatr. 2010, 78, 590–598. [Google Scholar] [CrossRef]
- Katano, H.; Fukushima, T.; Karasawa, K.; Sugiyama, N.; Ohkura, A.; Kamiya, K. Primidone-induced hyperammonemic encephalopathy in a patient with cerebral astrocytoma. J. Clin. Neurosci. 2002, 9, 79–81. [Google Scholar] [CrossRef]
- Sechi, G.; Murgia, B.; Sau, G.; Peddone, L.; Tirotto, A.; Barrocu, M.; Rosati, G. Asterixis and toxic encephalopathy induced by gabapentin. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 195–199. [Google Scholar] [CrossRef]
- Lemberg, A.; Fernández, M.A.; Coll, C.; Rosello, D.O.; Romay, S.; Perazzo, J.C.; Filinger, E.L. Reyes’s syndrome, encephalopathy, hyperammonemia and acetyl salicylic acid ingestion in a city hospital of Buenos Aires, Argentina. Curr. Drug Saf. 2009, 4, 17–21. [Google Scholar] [CrossRef]
- Kim, J.M.; Ryu, W.S.; Hwang, Y.H.; Kim, J.S. Aggravation of ataxia due to acetazolamide induced hyperammonaemia in episodic ataxia. J. Neurol. Neurosurg. Psychiatry 2007, 78, 771–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nott, L.; Price, T.J.; Pittman, K.; Patterson, K.; Fletcher, J. Hyperammonemia encephalopathy: An important cause of neurological deterioration following chemotherapy. Leuk. Lymphoma 2007, 48, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Lama, M.; Shannon, S.; Davin, Q. Methamphetamine Intoxication Encephalopathy Associated with Hyperammonemia. Psychosomatics 2016, 57, 325–329. [Google Scholar] [CrossRef]
- Crosbie, D.C.; Sugumar, H.; Simpson, M.A.; Walker, S.P.; Dewey, H.M.; Reade, M.C. Late-onset ornithine transcarbamylase deficiency: A potentially fatal yet treatable cause of coma. Crit. Care Resusc. 2009, 11, 222–227. [Google Scholar] [PubMed]
- Walker, V. Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes Obes. Metab. 2009, 11, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Shiohama, N.; Sugita, Y.; Imamura, N.; Sato, T.; Mizuno, Y. [Type II citrullinemia triggered by acetaminophen]. No Shinkei 1993, 45, 865–870. [Google Scholar]
- Summar, M.L.; Mew, N.A. Inborn Errors of Metabolism with Hyperammonemia: Urea Cycle Defects and Related Disorders. Pediatr. Clin. N. Am. 2018, 65, 231–246. [Google Scholar] [CrossRef]
- Ropper, A.H.; Samuels, M.A.; Klein, J.P.; Prasad, S. Adams & Victor’s Principles of Neurology, 11th ed.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Kleppe, S.; Mian, A.; Lee, B. Urea Cycle Disorders. Curr. Treat. Options Neurol. 2003, 5, 309–319. [Google Scholar] [CrossRef]
- Burton, B.K. Urea cycle disorders. Clin. Liver Dis. 2000, 4, 815–830. [Google Scholar] [CrossRef]
- Lichter-Konecki, U.; Caldovic, L.; Morizono, H.; Simpson, K.; Mew, N.A.; MacLeod, E.; Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; et al. Ornithine Transcarbamylase Deficiency. In Gene Reviews®; University of Seattle: Seattle, WA, USA, 1993–2021. [Google Scholar]
- Redant, S.; Empain, A.; Mugisha, A.; Kamgang, P.; Attou, R.; Honoré, P.M.; De Bels, D. Management of late onset urea cycle disorders-a remaining challenge for the intensivist? Ann. Intensive Care 2021, 11, 2. [Google Scholar] [CrossRef]
- Tuchman, M.; Plante, R.J.; McCann, M.T.; Qureshi, A.A. Seven new mutations in the human ornithine transcarbamylase gene. Hum. Mutat. 1994, 4, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.D.; Kostiner, D.R.; Weisiger, K.; Moffatt, E.; Lindeman, N.; Goodman, S.; Tuchman, M.; Packman, S. Acute fatal presentation of ornithine transcarbamylase deficiency in a previously healthy male. Hepatol. Int. 2008, 2, 390–394, Erratum in Hepatol. Int. 2008, 2, 395–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, K. Creutzfeldt-Jacob disease mimics, or how to sort out the subacute encephalopathy patient. Postgrad. Med. J. 2011, 87, 369–378. [Google Scholar] [CrossRef]
- Baddour, E.; Tewksbury, A.; Stauner, N. Valproic acid-induced hyperammonemia: Incidence, clinical significance, and treatment management. Ment. Health Clin. 2018, 8, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.N.; Marks, A.; Lizer, M.H. Carbamazepine-induced hyperammonemia. Am. J. Health Syst. Pharm. 2009, 66, 1468–1470. [Google Scholar] [CrossRef]
- Summar, M.L.; Dobbelaere, D.; Brusilow, S.; Lee, B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008, 97, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Stepien, K.M.; Geberhiwot, T.; Hendriksz, C.J.; Treacy, E.P. Challenges in diagnosing and managing adult patients with urea cycle disorders. J. Inherit. Metab. Dis. 2019, 42, 1136–1146. [Google Scholar] [CrossRef]
- Lin, C.; Tusa, J.K. Equimolar ammonia interference in potassium measurement on the Osmetech OPTI CCA: A reply. Clin. Chem. 2006, 52, 2116–2117. [Google Scholar] [CrossRef] [Green Version]
- Nøhr, T.K.; Eriksen, P.L.; Lund, A.; Vilstrup, H.; Thomsen, K.L. [Hyperammonaemic encephalopathy in adults without liver diseases]. Ugeskr Laeger. 2021, 183, V11200825. [Google Scholar]
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelle, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guideline for diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niclasen, B.H.; Schelde-Olesen, M.T.; Astvad, M.; Løkke, A.; Krøigård, T.; Nielsen, H.H. Hyperammonaemic Encephalopathy Caused by Adult-Onset Ornithine Transcarbamylase Deficiency. Brain Sci. 2022, 12, 231. https://doi.org/10.3390/brainsci12020231
Niclasen BH, Schelde-Olesen MT, Astvad M, Løkke A, Krøigård T, Nielsen HH. Hyperammonaemic Encephalopathy Caused by Adult-Onset Ornithine Transcarbamylase Deficiency. Brain Sciences. 2022; 12(2):231. https://doi.org/10.3390/brainsci12020231
Chicago/Turabian StyleNiclasen, Bjarke Hammer, Maria Therese Schelde-Olesen, Mads Astvad, Anders Løkke, Thomas Krøigård, and Helle H. Nielsen. 2022. "Hyperammonaemic Encephalopathy Caused by Adult-Onset Ornithine Transcarbamylase Deficiency" Brain Sciences 12, no. 2: 231. https://doi.org/10.3390/brainsci12020231