
Specific microRNAs for Modulation of Autophagy in Spinal Cord Injury



Abstract
:1. Introduction
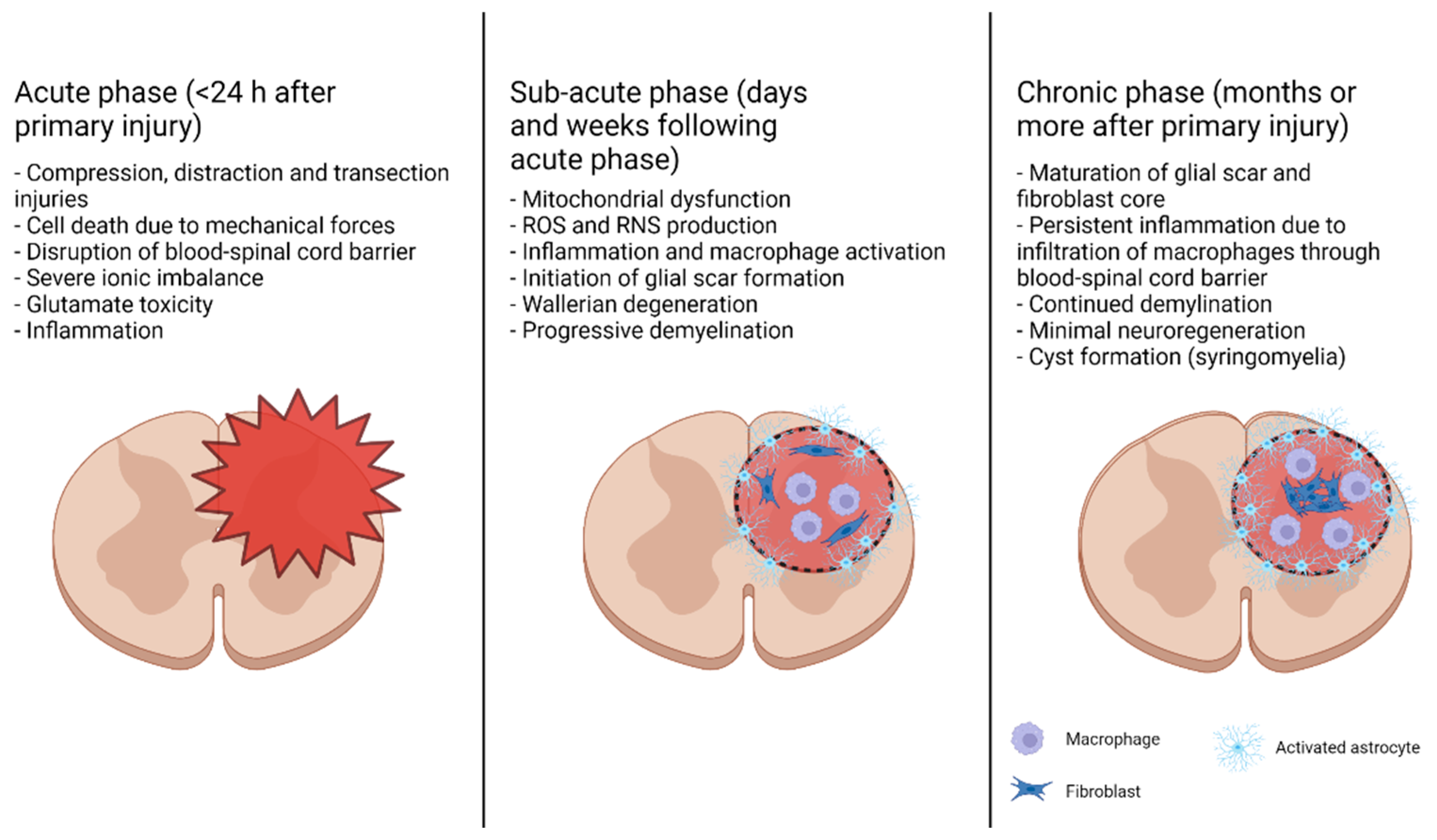
2. Different Phases of Traumatic SCI and Its Complex Pathophysiology
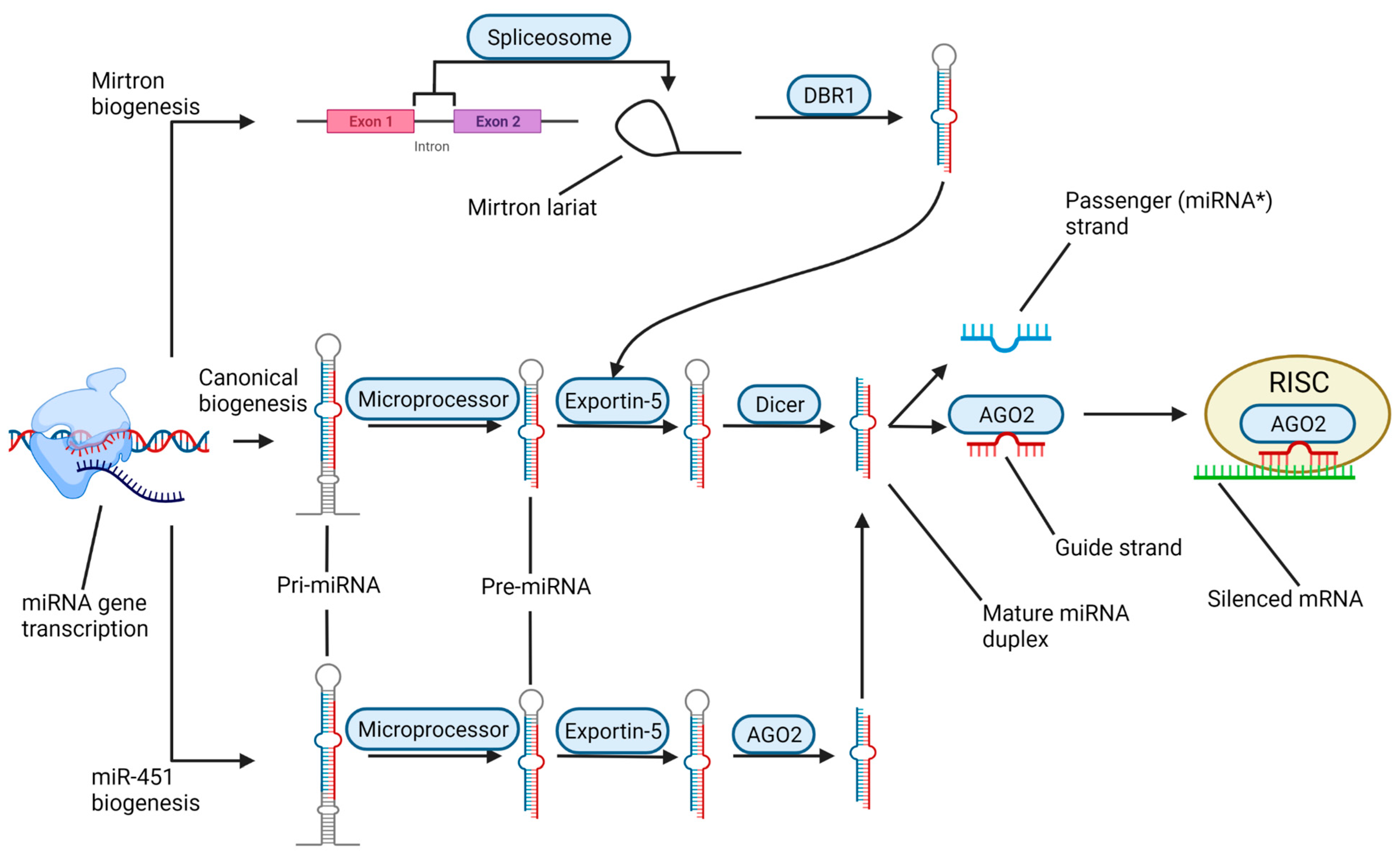
3. Biogenesis and Roles of miRNAs in the Context of SCI
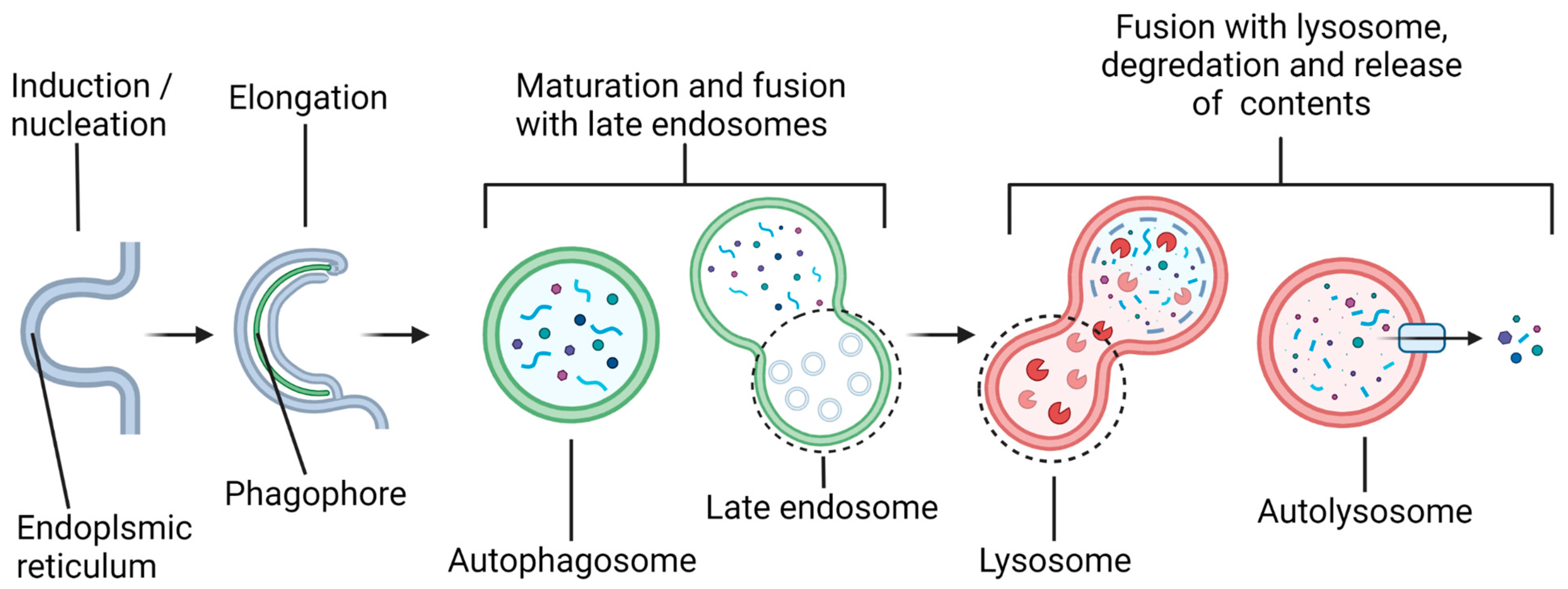
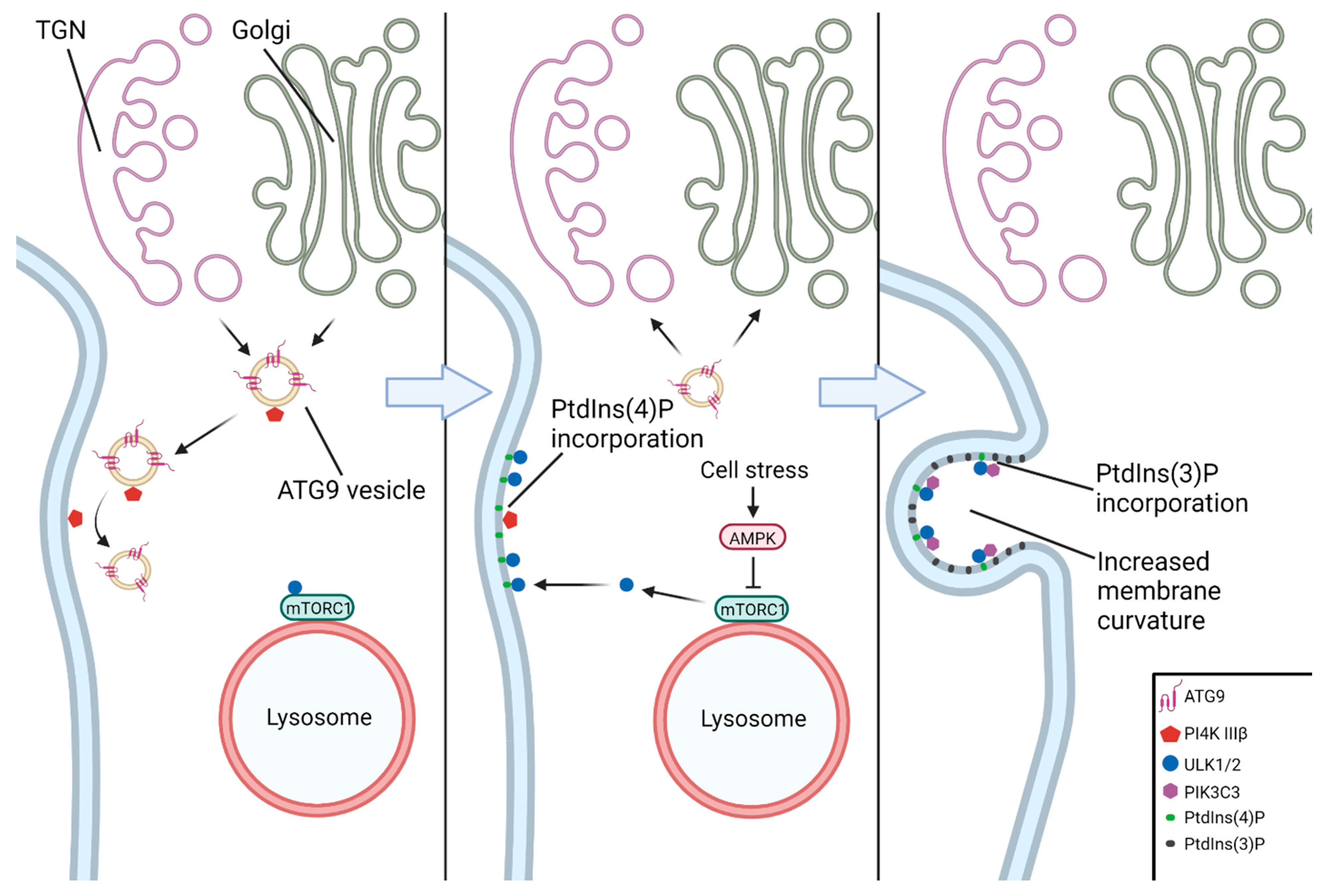
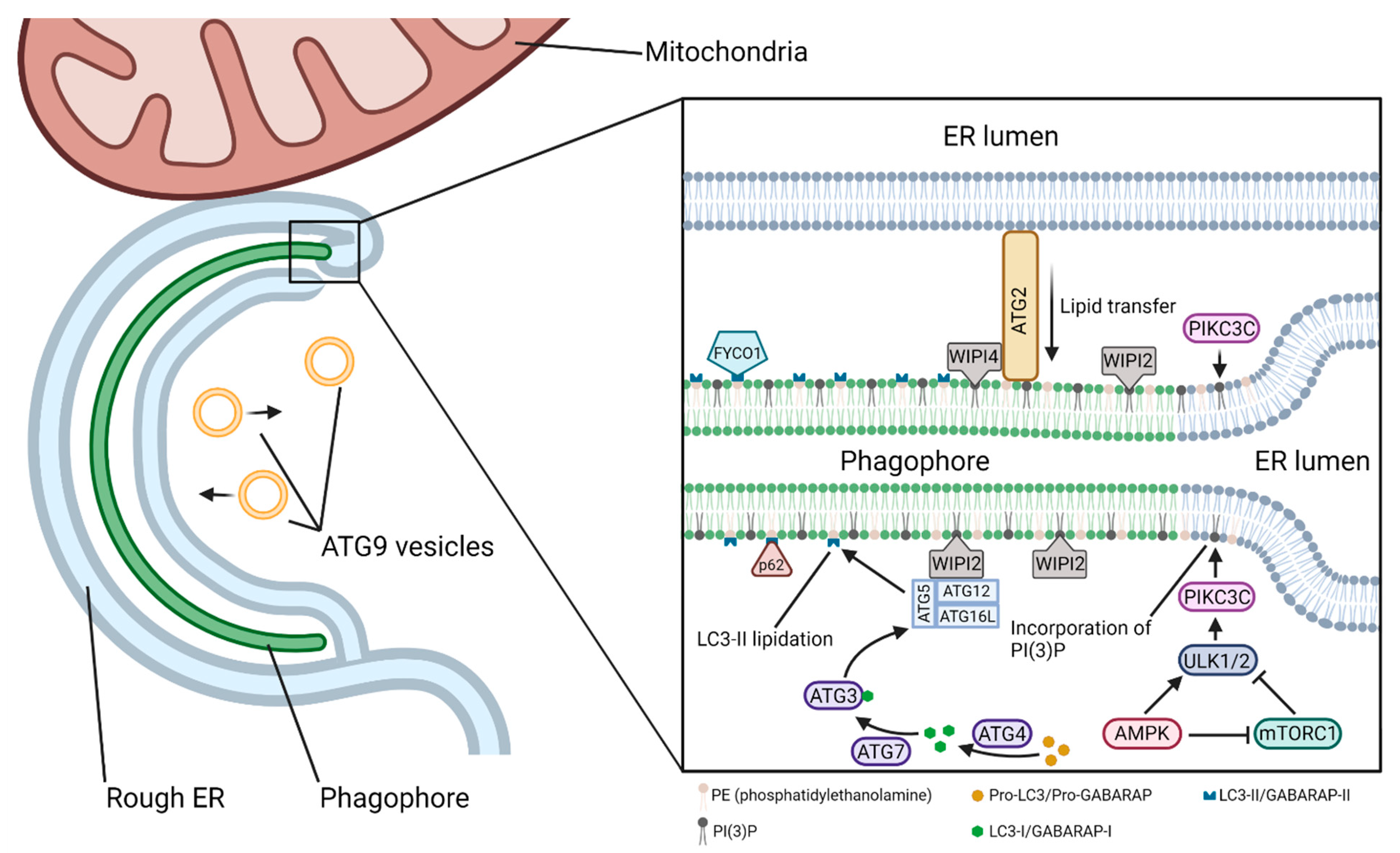
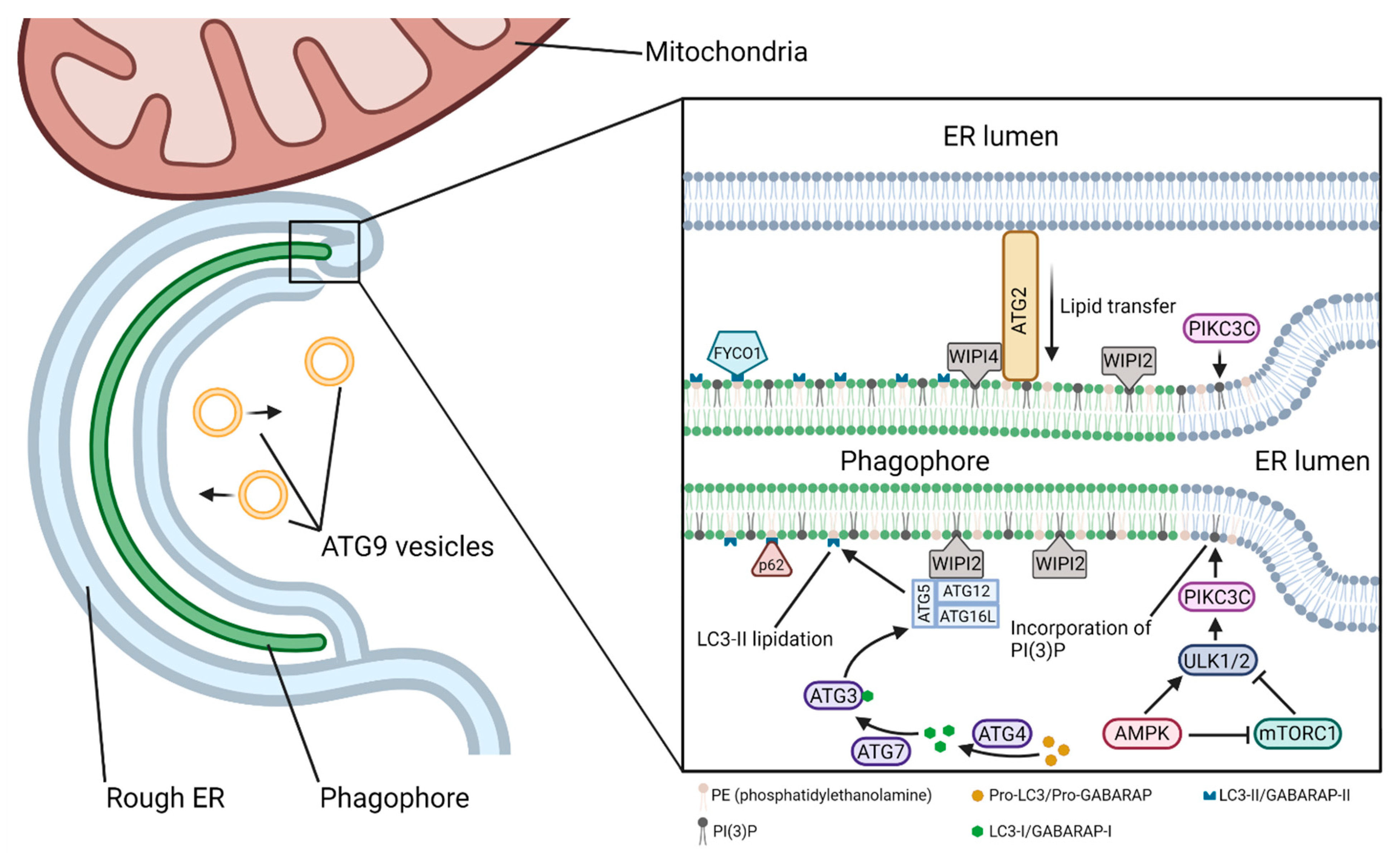
4. Components of the Autophagy Pathway from Nucleation to Fusion
5. Specific miRNAs in Modulation of Autophagy in Preclinical Models of SCI
5.1. Autophagy in Neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
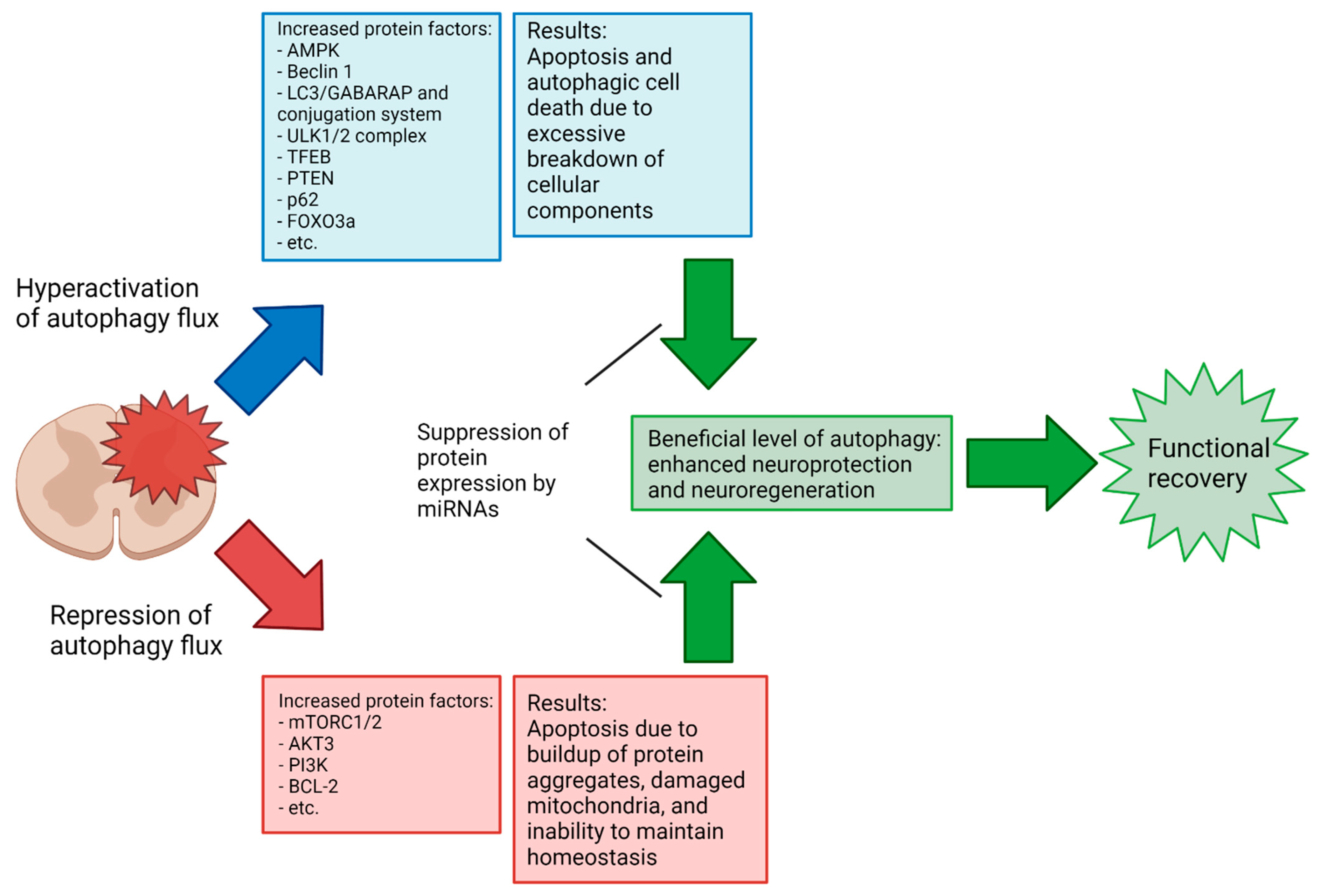{kind=link}
| microRNA | Molecular Target(s) | Effect on Autophagy Flux | Prospect of SCI Recovery | References |
|---|---|---|---|---|
| miR-93-5p | PTEN | Decrease | Beneficial | [115,116,117] |
| ATG7 | ||||
| TLR4 | ||||
| miR-384-5p | Beclin 1 | Decrease | Beneficial | [118,119] |
| GRP78 | ||||
| miR-378 | ATG12 | Tissue dependent | Beneficial | [120,121,122,123] |
| GRB2 | ||||
| miR-27a | FOXO3a | Decrease | Beneficial | [124,125,126,127] |
| DRAM2 | ||||
| PINK1 | ||||
| miR-223 | RPH1/KDM4A | Decrease | Beneficial | [128,129] |
| miR-124 | PI3K | Decrease | Beneficial | [130,131,132] |
| AMPK | ||||
| Bcl-2 | ||||
| p62 | ||||
| miR-212-3p | PTEN | Decrease | Beneficial | [133,134,135,136] |
| miR-15a | Akt3 | Increase | Beneficial (in neuropathic pain model) | [137,138] |
| Rictor | ||||
| miR-384-5p | Beclin 1 | Decrease | Beneficial | [139] |
| miR-223 | FOXO3a ATG16L | Decrease | Beneficial | [140,141,142,143,144] |
| miR-30 | Beclin 1 | Tissue dependent | Context dependent | [145,146,147,148,149] |
| miR-30d | Beclin 1 | Increase or decrease | Beneficial | [150,151,152] |
5.2. Autophagy in Glial Cells
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagy in the eukaryotic cell. Eukaryot. Cell 2002, 1, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Melia, T.J.; Lystad, A.H.; Simonsen, A. Autophagosome biogenesis: From membrane growth to closure. J. Cell Biol. 2020, 219, e202002085. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and autophagy: Interactions and molecular regulatory mechanisms. Cell Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in human diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 23–34. [Google Scholar] [CrossRef]
- Zhou, K.; Sansur, C.A.; Xu, H.; Jia, X. The temporal pattern, flux, and function of autophagy in spinal cord injury. Int. J. Mol. Sci. 2017, 18, 466. [Google Scholar] [CrossRef] [Green Version]
- Lo, J.; Chan, L.; Flynn, S. A systematic review of the incidence, prevalence, costs, and activity and work limitations of amputation, osteoarthritis, rheumatoid arthritis, back pain, multiple sclerosis, spinal cord injury, stroke, and traumatic brain injury in the United States: A 2019 update. Arch. Phys. Med. Rehabil. 2021, 102, 115–131. [Google Scholar]
- Khorasanizadeh, M.; Yousefifard, M.; Eskian, M.; Lu, Y.; Chalangari, M.; Harrop, J.S.; Jazayeri, S.B.; Seyedpour, S.; Khodaei, B.; Hosseini, M.; et al. Neurological recovery following traumatic spinal cord injury: A systematic review and meta-analysis. J. Neurosurg. Spine 2019, 130, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, C.S.; Wilson, J.R.; Nori, S.; Kotter, M.R.N.; Druschel, C.; Curt, A.; Fehlings, M.G. Traumatic spinal cord injury. Nat. Rev. Dis. Primers 2017, 3, 17018. [Google Scholar] [CrossRef] [PubMed]
- Flack, J.A.; Sharma, K.D.; Xie, J.Y. Delving into the recent advancements of spinal cord injury treatment: A review of recent progress. Neural Regen. Res. 2022, 17, 283–291. [Google Scholar] [PubMed]
- Hejrati, N.; Fehlings, M.G. A review of emerging neuroprotective and neuroregenerative therapies in traumatic spinal cord injury. Curr. Opin. Pharmacol. 2021, 60, 331–340. [Google Scholar] [CrossRef]
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic spinal cord injury: An overview of pathophysiology, models and acute injury mechanisms. Front. Neurol. 2019, 10, 282. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, C.S.; Nori, S.; Tetreault, L.; Wilson, J.; Kwon, B.; Harrop, J.; Choi, D.; Fehlings, M.G. Traumatic spinal cord injury-repair and regeneration. Neurosurgery 2017, 80, S9–S22. [Google Scholar] [CrossRef]
- Anjum, A.; Yazid, M.D.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Kumar, R.K.A.; Lokanathan, Y. Spinal cord injury: Pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int. J. Mol. Sci. 2020, 21, 7533. [Google Scholar] [CrossRef]
- Hayta, E.; Elden, H. Acute spinal cord injury: A review of pathophysiology and potential of non-steroidal anti-inflammatory drugs for pharmacological intervention. J. Chem. Neuroanat. 2018, 87, 25–31. [Google Scholar] [CrossRef]
- Tran, A.P.; Warren, P.M.; Silver, J. The biology of regeneration failure and success after spinal cord injury. Physiol. Rev. 2018, 98, 881–917. [Google Scholar] [CrossRef]
- Roy, A.; Pathak, Z.; Kumar, H. Strategies to neutralize RhoA/ROCK pathway after spinal cord injury. Exp. Neurol. 2021, 343, 113794. [Google Scholar] [CrossRef]
- Dyck, S.M.; Alizadeh, A.; Santhosh, K.T.; Proulx, E.H.; Wu, C.-L.; Karimi-Abdolrezaee, S. Chondroitin sulfate proteoglycans negatively modulate spinal cord neural precursor cells by signaling through LAR and RPTPσ and modulation of the Rho/ROCK pathway. Stem Cells 2015, 33, 2550–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, K.D.; Nguyen, H.X.; Galvan, M.D.; Salazar, D.L.; Woodruff, T.M.; Anderson, A.J. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010, 133, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaumt, R.L.; Ambrost, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The metazoan microRNA complement. Nucleic Acids Res. 2020, 48, D132–D141. [Google Scholar] [CrossRef] [Green Version]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.-P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Talukder, A.; Cha, M.; Li, X.; Hu, H. Computational annotation of miRNA transcription start sites. Brief Bioinform. 2021, 22, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.A.; Jo, M.H.; Choi, Y.-G.; Park, J.; Kwon, S.C.; Hohng, S.; Kim, V.N.; Woo, J.-S. Functional anatomy of the human microprocessor. Cell 2015, 161, 1374–1387. [Google Scholar] [CrossRef] [Green Version]
- Stavast, C.J.; Erkeland, S.J. The non-canonical aspects of microRNAs: Many roads to gene regulation. Cells 2019, 8, 1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Medley, J.C.; Panzade, G.; Zinovyeva, A.Y. microRNA strand selection: Unwinding the rules. Wiley Interdiscip. Rev. RNA 2021, 12, e1627. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. microRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Rong, Y.; Ji, C.; Lv, C.; Jiang, D.; Ge, X.; Gong, F.; Tang, P.; Cai, W.; Liu, W.; et al. microRNA-421-3p-abundant small extracellular vesicles derived from M2 bone marrow-derived macrophages attenuate apoptosis and promote motor function recovery via inhibition of mTOR in spinal cord injury. J. Nanobiotechnol. 2020, 18, 72. [Google Scholar] [CrossRef]
- Corona Velazquez Angel, F.; Jackson William, T. So many roads: The multifaceted regulation of autophagy induction. Mol. Cell Biol. 2018, 38, e00303-18. [Google Scholar] [CrossRef] [Green Version]
- Anwar, T.; Liu, X.; Suntio, T.; Marjamäki, A.; Biazik, J.; Chan, E.Y.W.; Varjosalo, M.; Eskelinen, E.-L. ER-targeted Beclin 1 supports autophagosome biogenesis in the absence of ULK1 and ULK2 kinases. Cells 2019, 8, 475. [Google Scholar] [CrossRef] [Green Version]
- McAlpine, F.; Williamson, L.E.; Tooze, S.A.; Chan, E.Y.W. Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy 2013, 9, 361–373. [Google Scholar] [CrossRef]
- Wang, B.; Kundu, M. Canonical and noncanonical functions of ULK/Atg1. Curr. Opin. Cell Biol. 2017, 45, 47–54. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Hall, M.N.; Lin, S.-C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogala, K.B.; Gu, X.; Kedir, J.F.; Abu-Remaileh, M.; Bianchi, L.F.; Bottino, A.M.S.; Dueholm, R.; Niehaus, A.; Overwijn, D.; Fils, A.-C.P.; et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 2019, 366, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, K.; Tokunaga, C.; Oshiro, N.; Yoshino, K.-I. Raptor, a binding partner of target of rapamycin. Biochem. Biophys. Res. Commun. 2004, 313, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [PubMed] [Green Version]
- Van Nostrand, J.L.; Hellberg, K.; Luo, E.-C.; Van Nostrand, E.L.; Dayn, A.; Yu, J.; Shokhirev, M.N.; Dayn, Y.; Yeo, G.W.; Shaw, R.J. AMPK regulation of Raptor and TSC2 mediate metformin effects on transcriptional control of anabolism and inflammation. Genes Dev. 2020, 34, 1330–1344. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Paquette, M.; El-Houjeiri, L.; C Zirden, L.; Puustinen, P.; Blanchette, P.; Jeong, H.; Dejgaard, K.; Siegel, P.M.; Pause, A. AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy 2021, 17, 3957–3975. [Google Scholar] [CrossRef]
- Löffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Seo, M.; Otto, N.M.; Kim, D.-H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 2011, 7, 1212–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Karanasios, E.; Walker, S.A.; Okkenhaug, H.; Manifava, M.; Hummel, E.; Zimmermann, H.; Ahmed, Q.; Domart, M.; Collinson, L.; Ktistakis, N.T. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 2016, 7, 12420. [Google Scholar] [CrossRef] [PubMed]
- Judith, D.; Jefferies, H.B.J.; Boeing, S.; Frith, D.; Snijders, A.P.; Tooze, S.A. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J. Cell Biol. 2019, 218, 1634–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanasios, E.; Stapleton, E.; Manifava, M.; Kaizuka, T.; Mizushima, N.; Walker, S.A.; Ktistakis, N.T. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 2013, 126, 5224–5238. [Google Scholar] [CrossRef] [Green Version]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef]
- Chen, Y.-D.; Fang, Y.-T.; Chang, C.-P.; Lin, C.-F.; Hsu, L.-J.; Wu, S.-R.; Chiu, Y.-C.; Anderson, R.; Lin, Y.-S. S100A10 regulates ULK1 localization to ER–mitochondria contact sites in IFN-γ-triggered autophagy. J. Mol. Biol. 2017, 429, 142–157. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Giordano, F.; Dupont, N.; Grasso, D.; Vaccaro, M.I.; Codogno, P.; Morel, E. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 2017, 36, 2018–2033. [Google Scholar] [CrossRef]
- Kohler, V.; Aufschnaiter, A.; Büttner, S. Closing the gap: Membrane contact sites in the regulation of autophagy. Cells 2020, 9, 1184. [Google Scholar] [CrossRef]
- Proikas-Cezanne, T.; Takacs, Z.; Dönnes, P.; Kohlbacher, O. WIPI proteins: Essential PtdIns3P effectors at the nascent autophagosome. J. Cell Sci. 2015, 128, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Bakula, D.; Müller, A.J.; Zuleger, T.; Takacs, Z.; Franz-Wachtel, M.; Thost, A.-K.; Brigger, D.; Tschan, M.P.; Frickey, T.; Robenek, H.; et al. WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat. Commun. 2017, 8, 15637. [Google Scholar] [CrossRef] [PubMed]
- Strong, L.M.; Chang, C.; Riley, J.F.; Boecker, C.A.; Flower, T.G.; Buffalo, C.Z.; Ren, X.; Stavoe, A.K.; Holzbaur, E.L.; Hurley, J.H. Structural basis for membrane recruitment of ATG16L1 by WIPI2 in autophagy. Elife 2021, 10, e70372. [Google Scholar] [CrossRef] [PubMed]
- Lystad, A.H.; Simonsen, A. Phosphoinositide-binding proteins in autophagy. FEBS Lett. 2016, 590, 2454–2468. [Google Scholar] [CrossRef] [PubMed]
- Polson, H.E.J.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lystad, A.H.; Simonsen, A. Mechanisms and pathophysiological roles of the ATG8 conjugation machinery. Cells 2019, 8, 973. [Google Scholar] [CrossRef] [Green Version]
- Johansen, T.; Lamark, T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Biazik, J.; Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, N.; Chauhan, S.; Arko-Mensah, J.; Castillo, E.F.; Masedunskas, A.; Weigert, R.; Robenek, H.; Proikas-Cezanne, T.; Deretic, V. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr. Biol. 2014, 24, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Gómez-Sánchez, R.; Rose, J.; Guimarães, R.; Mari, M.; Papinski, D.; Rieter, E.; Geerts, W.J.; Hardenberg, R.; Kraft, C.; Ungermann, C.; et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J. Cell Biol. 2018, 217, 2743–2763. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schütter, M.; Giavalisco, P.; Brodesser, S.; Graef, M. Local fatty acid channeling into phospholipid synthesis drives phagophore expansion during autophagy. Cell 2020, 180, 135–149.e14. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Gowan, S.; Lin, G.; Wong, T.; Fong, A.-C.L.; Shamsaei, E.; Parkes, H.G.; Mui, J.; Raynaud, F.; Asad, Y.; et al. De novo phosphatidylcholine synthesis is required for autophagosome membrane formation and maintenance during autophagy. Autophagy 2020, 16, 1044–1060. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Huang, W.; Wang, W. Multifaceted roles of COPII subunits in autophagy. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118627. [Google Scholar] [CrossRef]
- Staiano, L.; Zappa, F. Hijacking intracellular membranes to feed autophagosomal growth. FEBS Lett. 2019, 593, 3120–3134. [Google Scholar] [CrossRef]
- Puri, C.; Vicinanza, M.; Rubinsztein, D.C. Phagophores evolve from recycling endosomes. Autophagy 2018, 14, 1475–1477. [Google Scholar] [CrossRef]
- Maeda, S.; Yamamoto, H.; Kinch, L.N.; Garza, C.M.; Takahashi, S.; Otomo, C.; Grishin, N.V.; Forli, S.; Mizushima, N.; Otomo, T. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat. Struct. Mol. Biol. 2020, 27, 1194–1201. [Google Scholar] [CrossRef]
- Zhen, Y.; Spangenberg, H.; Munson, M.J.; Brech, A.; Schink, K.O.; Tan, K.-W.; Sørensen, V.; Wenzel, E.M.; Radulovic, M.; Engedal, N.; et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 2020, 16, 826–841. [Google Scholar] [CrossRef] [Green Version]
- Djeddi, A.; Michelet, X.; Culetto, E.; Alberti, A.; Barois, N.; Legouis, R. Induction of autophagy in ESCRT mutants is an adaptive response for cell survival in C. elegans. J. Cell Sci. 2012, 125, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Oshima, R.; Hasegawa, T.; Tamai, K.; Sugeno, N.; Yoshida, S.; Kobayashi, J.; Kikuchi, A.; Baba, T.; Futatsugi, A.; Sato, I.; et al. ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci. Rep. 2016, 6, 24997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamai, K.; Tanaka, N.; Nara, A.; Yamamoto, A.; Nakagawa, I.; Yoshimori, T.; Ueno, Y.; Shimosegawa, T.; Sugamura, K. Role of Hrs in maturation of autophagosomes in mammalian cells. Biochem. Biophys. Res. Commun. 2007, 360, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Liang, X.; Hattori, T.; Tang, Z.; He, H.; Chen, H.; Liu, X.; Abraham, T.; Imamura-Kawasawa, Y.; Buchkovich, N.J.; et al. VPS37A directs ESCRT recruitment for phagophore closure. J. Cell Biol. 2019, 218, 3336–3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Wu, Z.; Zhao, M.; Murtazina, R.; Cai, J.; Zhang, A.; Li, R.; Sun, D.; Li, W.; Zhao, L.; et al. Rab5-dependent autophagosome closure by ESCRT. J. Cell Biol. 2019, 218, 1908–1927. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Cheng, S.; Zhao, H.; Zou, W.; Yoshina, S.; Mitani, S.; Zhang, H.; Wang, X. PI3P phosphatase activity is required for autophagosome maturation and autolysosome formation. EMBO Rep. 2014, 15, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.-Q.; Ni, T.; Hong, B.; Wang, H.-Y.; Jiang, F.-J.; Zou, S.; Chen, Y.; Zheng, X.-L.; Klionsky, D.J.; Liang, Y.; et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy 2012, 8, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef]
- Ganesan, D.; Cai, Q. Understanding amphisomes. Biochem. J. 2021, 478, 1959–1976. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Lőrincz, P.; Juhász, G. Autophagosome-lysosome fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef] [PubMed]
- Ebner, P.; Poetsch, I.; Deszcz, L.; Hoffmann, T.; Zuber, J.; Ikeda, F. The IAP family member BRUCE regulates autophagosome-lysosome fusion. Nat. Commun. 2018, 9, 599. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.-E.; Cheung, Y.W.S.; Nguyen, T.N.; Gong, M.; Chan, S.; Lazarou, M.; Yip, C.K. Insights on autophagosome-lysosome tethering from structural and biochemical characterization of human autophagy factor EPG5. Commun. Biol. 2021, 4, 291. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zheng, P.; Zhou, C.; Wang, X.; Ma, H.; Ma, W.; Zhou, X.; Teng, J.; Chen, J. DIPK2A promotes STX17- and VAMP7-mediated autophagosome-lysosome fusion by binding to VAMP7B. Autophagy 2020, 16, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Jiang, P.; Nakano, S.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 2018, 217, 2633–2645. [Google Scholar] [CrossRef]
- Tang, B.L. Syntaxin 16′s newly deciphered roles in autophagy. Cells 2019, 8, 1655. [Google Scholar] [CrossRef] [Green Version]
- Almurshidi, B.; Carver, W.; Scott, G.; Ray, S.K. Roles of miRNAs in spinal cord injury and potential therapeutic interventions. Neuroimmunol. Neuroinflamm. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zheng, W.; Dai, L.; Fu, S.; Shi, E. Bone marrow mesenchymal stem cell derived exosomal miR-455-5p protects against spinal cord ischemia reperfusion injury. Tissue Cell 2022, 74, 101678. [Google Scholar] [CrossRef]
- Bisicchia, E.; Sasso, V.; Catanzaro, G.; Leuti, A.; Besharat, Z.M.; Chiacchiarini, M.; Molinari, M.; Ferretti, E.; Viscomi, M.T.; Chiurchiù, V. Resolvin D1 halts remote neuroinflammation and improves functional recovery after focal brain damage Via ALX/FPR2 receptor-regulated microRNAs. Mol. Neurobiol. 2018, 55, 6894–6905. [Google Scholar] [CrossRef] [Green Version]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Saleem, S. Apoptosis, autophagy, necrosis and their multi galore crosstalk in neurodegeneration. Neuroscience 2021, 469, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Dong, X.; Zhao, R.; Zhang, R.; Xu, C.; Wang, X.; Liu, C.; Hu, X.; Huang, S.; Chen, L. Cadmium results in accumulation of autophagosomes-dependent apoptosis through activating Akt-impaired autophagic flux in neuronal cells. Cell Signal. 2019, 55, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.R.; Cramer, S.D.; Thorburn, A. The interplay of autophagy and non-apoptotic cell death pathways. Int. Rev. Cell Mol. Biol. 2020, 352, 159–187. [Google Scholar]
- Ray, S.K. Modulation of autophagy for neuroprotection and functional recovery in traumatic spinal cord injury. Neural Regen. Res. 2020, 15, 1601–1612. [Google Scholar] [CrossRef]
- Fang, B.; Li, X.-Q.; Bao, N.-R.; Tan, W.-F.; Chen, F.-S.; Pi, X.-L.; Zhang, Y.; Ma, H. Role of autophagy in the bimodal stage after spinal cord ischemia reperfusion injury in rats. Neuroscience 2016, 328, 107–116. [Google Scholar] [CrossRef]
- Zhang, D.; Zhu, D.; Wang, F.; Zhu, J.-C.; Zhai, X.; Yuan, Y.; Li, C.-X. Therapeutic effect of regulating autophagy in spinal cord injury: A network meta-analysis of direct and indirect comparisons. Neural Regen. Res. 2020, 15, 1120–1132. [Google Scholar] [CrossRef]
- Valencia, M.; Kim, S.R.; Jang, Y.; Lee, S.H. Neuronal autophagy: Characteristic features and roles in neuronal pathophysiology. Biomol. Ther. 2021, 29, 605–614. [Google Scholar] [CrossRef]
- Roney, J.C.; Li, S.; Farfel-Becker, T.; Huang, N.; Sun, T.; Xie, Y.; Cheng, X.-T.; Lin, M.-Y.; Platt, F.M.; Sheng, Z.-H. Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type, C. Dev. Cell 2021, 56, 1452–1468.e8. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.; Azarnia Tehran, D.; Haucke, V.; Soykan, T. The axonal endolysosomal and autophagic systems. J. Neurochem. 2021, 158, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Holzbaur, E.L.F. Compartment-specific regulation of autophagy in primary neurons. J. Neurosci. 2016, 36, 5933–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariosa, A.R.; Klionsky, D.J. Autophagy core machinery: Overcoming spatial barriers in neurons. J. Mol. Med. 2016, 94, 1217–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Jin, Y.; Li, Q.; Sun, X.; Zhu, H.; Cui, H. MiR-93-5p targeting PTEN regulates the NMDA-induced autophagy of retinal ganglion cells via AKT/mTOR pathway in glaucoma. Biomed. Pharmacother. 2018, 100, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, Y.; Liang, X.; Xing, X.; Xu, X.; Zhou, C. Toll-like receptor 4 knockdown attenuates brain damage and neuroinflammation after traumatic brain injury via inhibiting neuronal autophagy and astrocyte activation. Cell Mol. Neurobiol. 2018, 38, 1009–1019. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, M.; Deng, S.; Lu, J.; Huang, H.; Zhang, Y.; Gong, P.; Shen, X.; Ruan, H.; Jin, M.; et al. miR-93-5p-Containing exosomes treatment attenuates acute myocardial infarction-induced myocardial damage. Mol. Ther. Nucleic Acids 2018, 11, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhong, Y.; Huang, D.; Li, J. Macrophage autophagy regulated by miR-384-5p-mediated control of Beclin-1 plays a role in the development of atherosclerosis. Am. J. Transl. Res. 2016, 8, 606–614. [Google Scholar]
- Zhou, Z.; Hu, B.; Lyu, Q.; Xie, T.; Wang, J.; Cai, Q. miR-384-5p promotes spinal cord injury recovery in rats through suppressing of autophagy and endoplasmic reticulum stress. Neurosci. Lett. 2020, 727, 134937. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, H.; Yang, H.; Zhan, Y.; Liu, X. miR-378-3p alleviates contusion spinal cord injury by negatively regulating ATG12. Int. J. Exp. Pathol. 2021, 102, 200–208. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, F.; Ma, W.; Zhang, P. Suppression of long noncoding RNA NEAT1 attenuates hypoxia-induced cardiomyocytes injury by targeting miR-378a-3p. Gene 2020, 731, 144324. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.-C.; Yi, T.-Z.; Huang, F.-G.; Wei, Y.; Luo, X.-P.; Luo, Q.-S. Role of long noncoding RNA MEG3/miR-378/GRB2 axis in neuronal autophagy and neurological functional impairment in ischemic stroke. J. Biol. Chem. 2020, 295, 14125–14139. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, J.; Liu, W.; Wang, H.; Zhao, L.; Liu, S.; Li, P.; Zhang, S.; Sun, C.; Wu, Y.; et al. microRNA-378 promotes autophagy and inhibits apoptosis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2018, 115, E10849–E10858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Liao, W.; Yang, J.; Ma, K.; Li, X.; Wang, Y.; Wang, D.; Wang, L.; Zhang, Y.; Yin, Y.; et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 2012, 8, 1712–1723. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhao, M.; Wang, Y.; Liu, A.; Lv, M.; Li, Y.; Yang, X.; Wu, Z. Neuroprotective effects of miR-27a against traumatic brain injury via suppressing FoxO3a-mediated neuronal autophagy. Biochem. Biophys. Res. Commun. 2017, 482, 1141–1147. [Google Scholar] [CrossRef]
- Li, H.; Lu, C.; Yao, W.; Xu, L.; Zhou, J.; Zheng, B. Dexmedetomidine inhibits inflammatory response and autophagy through the circLrp1b/miR-27a-3p/Dram2 pathway in a rat model of traumatic brain injury. Aging 2020, 12, 21687–21705. [Google Scholar] [CrossRef]
- Kim, J.; Fiesel, F.C.; Belmonte, K.C.; Hudec, R.; Wang, W.-X.; Kim, C.; Nelson, P.T.; Springer, W.; Kim, J. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Niu, Y.; Li, D.; Zhang, Q. microRNA-223 alleviates lipopolysaccharide-induced PC-12 cells apoptosis and autophagy by targeting RPH1 in spinal cord injury. Int. J. Clin. Exp. Pathol. 2017, 10, 9223–9232. [Google Scholar]
- Bernard, A.; Jin, M.; González-Rodríguez, P.; Füllgrabe, J.; Delorme-Axford, E.; Backues, S.K.; Joseph, B.; Klionsky, D.J. Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy. Curr. Biol. 2015, 25, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Feng, Z.; Du, L.; Huang, Y.; Ge, J.; Deng, Y.; Mei, Z. The potential role of microRNA-124 in cerebral ischemia injury. Int. J. Mol. Sci. 2019, 21, 120. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Yan, L.; Jiang, X.; Yu, Y.; Liu, H.; Gu, T.; Shi, E. Acquired inhibition of microRNA-124 protects against spinal cord ischemia-reperfusion injury partially through a mitophagy-dependent pathway. J. Thorac. Cardiovasc. Surg. 2017, 154, 1498–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, W.; Yan, Y.; Bao, T.-H.; Jia, W.-J.; Yang, F.; Wang, Y.; Zhu, Y.; Yin, M.; Han, J. Ischemic postconditioning exerts neuroprotective effect through negatively regulating PI3K/Akt2 signaling pathway by microRNA-124. Biomed. Pharmacother. 2020, 126, 109786. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Luan, L.; Li, J.; Yang, L. miR-212-3p improves rat functional recovery and inhibits neurocyte apoptosis in spinal cord injury models via PTEN downregulation-mediated activation of AKT/mTOR pathway. Brain Res. 2021, 1768, 147576. [Google Scholar] [CrossRef] [PubMed]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Kim, E.H.; Lee, Y.-J.; Sai, S.; Lim, S.H.; Park, J.W.; Chung, H.K.; Kim, J.; Vares, G.; Takahashi, A.; et al. Synergistic autophagy effect of miR-212-3p in zoledronic acid-treated in vitro and orthotopic in vivo models and in patient-derived osteosarcoma cells. Cancers 2019, 11, 1812. [Google Scholar] [CrossRef] [Green Version]
- Ramalinga, M.; Roy, A.; Srivastava, A.; Bhattarai, A.; Harish, V.; Suy, S.; Collins, S.; Kumar, D. microRNA-212 negatively regulates starvation induced autophagy in prostate cancer cells by inhibiting SIRT1 and is a modulator of angiogenesis and cellular senescence. Oncotarget 2015, 6, 34446–34457. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Liu, X.; Guo, Q.; Huang, Q.; Zhang, Q.; Cao, Z. miR-15a attenuates peripheral nerve injury-induced neuropathic pain by targeting AKT3 to regulate autophagy. Genes Genom. 2020, 42, 77–85. [Google Scholar] [CrossRef]
- Huang, N.; Wu, J.; Qiu, W.; Lyu, Q.; He, J.; Xie, W.; Xu, N.; Zhang, Y. miR-15a and miR-16 induce autophagy and enhance chemosensitivity of camptothecin. Cancer Biol. Ther. 2015, 16, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Huang, J.; Li, J.; Zhong, Y. Control of macrophage autophagy by miR-384-5p in the development of diabetic encephalopathy. Am. J. Transl. Res. 2018, 10, 511–518. [Google Scholar]
- Li, Y.; Zhou, D.; Ren, Y.; Zhang, Z.; Guo, X.; Ma, M.; Xue, Z.; Lv, J.; Liu, H.; Xi, Q.; et al. miR-223 restrains autophagy and promotes CNS inflammation by targeting ATG16L1. Autophagy 2019, 15, 478–492. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Chen, H.; Zhong, Y.; Yang, Q.; Wang, X.; Chen, R.; Guo, Y. microRNA-223 targeting ATG16L1 affects microglial autophagy in the kainic acid model of temporal lobe epilepsy. Front. Neurol. 2021, 12, 704550. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, X.; Cui, X.; Kuang, W.; Li, D.; Wang, J. Quercetin prevents isoprenaline-induced myocardial fibrosis by promoting autophagy via regulating miR-223-3p/FOXO3. Cell Cycle 2021, 20, 1253–1269. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Cen, S.; Zhong, Z.; Zhou, C.; Zhong, G. FOXO3 is targeted by miR-223-3p and promotes osteogenic differentiation of bone marrow mesenchymal stem cells by enhancing autophagy. Hum. Cell. 2021, 34, 14–27. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, E.; Tang, Y.; Mao, J.; Shen, J.; Zheng, X.; Xie, S.; Zhang, S.; Wu, Y.; Liu, H.; et al. miR-223 overexpression inhibits doxorubicin-induced autophagy by targeting FOXO3a and reverses chemoresistance in hepatocellular carcinoma cells. Cell Death Dis. 2019, 10, 843. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Liang, J.; Li, Y.; Li, J.; Yang, X.; Zhang, X.; Han, S.; Li, S.; Li, J. Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem. Res. 2014, 39, 1279–1291. [Google Scholar] [CrossRef]
- Sun, B.; Ou, H.; Ren, F.; Guan, Y.; Huan, Y.; Cai, H. Propofol protects against cerebral ischemia/reperfusion injury by down-regulating long noncoding RNA SNHG14. ACS Chem. Neurosci. 2021, 12, 3002–3014. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, H.-K.; Li, Y.-P.; Guo, Y.-P. Hydrogen sulfide protects spinal cord and induces autophagy via miR-30c in a rat model of spinal cord ischemia-reperfusion injury. J. Biomed. Sci. 2015, 22, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhong, X.; Tanyi, J.L.; Shen, J.; Xu, C.; Gao, P.; Zheng, T.M.; DeMichele, A.; Zhang, L. miR-30d Regulates multiple genes in the autophagy pathway and impairs autophagy process in human cancer cells. Biochem. Biophys. Res. Commun. 2013, 431, 617–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, M.; Klionsky, D.J.; Ray, S.K. miR-30e Blocks autophagy and acts synergistically with proanthocyanidin for inhibition of AVEN and BIRC6 to increase apoptosis in glioblastoma stem cells and glioblastoma SNB19 Cells. PLoS ONE 2016, 11, e0158537. [Google Scholar] [CrossRef]
- Zhao, F.; Qu, Y.; Wang, H.; Huang, L.; Zhu, J.; Li, S.; Tong, Y.; Zhang, L.; Li, J.; Mu, D. The effect of miR-30d on apoptosis and autophagy in cultured astrocytes under oxygen-glucose deprivation. Brain Res. 2017, 1671, 67–76. [Google Scholar] [CrossRef]
- Zhao, F.; Qu, Y.; Zhu, J.; Zhang, L.; Huang, L.; Liu, H.; Li, S.; Mu, D. miR-30d-5p Plays an important role in autophagy and apoptosis in developing rat brains after hypoxic-ischemic Injury. J. Neuropathol. Exp. Neurol. 2017, 76, 709–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Wang, H.; Jin, M.; Yang, X.; Ji, H.; Jiang, Y.; Zhang, H.; Wu, F.; Wu, G.; Lai, X.; et al. Exosomes from miR-30d-5p-adscs reverse acute ischemic stroke-induced, autophagy-mediated brain injury by promoting M2 microglial/macrophage polarization. Cell Physiol. Biochem. 2018, 47, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Guohui, M.; Li, D.; Bai, F.; Fang, J.; Zhang, G.; Xing, Y.; Zhou, J.; Guo, Y.; Kan, Y. lncRNA C2dat2 facilitates autophagy and apoptosis via the miR-30d-5p/DDIT4/mTOR axis in cerebral ischemia-reperfusion injury. Aging 2021, 13, 11315–11335. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.D.; He, X.G.; Yang, F.G.; Liu, M.Q.; Wang, Y.D.; Zhu, D.X.; Zhang, G.Z.; Ma, Z.J.; Kang, X.W. Research progress on the regulatory role of microRNAs in spinal cord injury. Regen Med. 2021, 16, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Ahn, J.; Hebron, M.; Chiu, T.; Ayoub, R.; Mulki, S.; Ressom, H.; Torres-Yaghi, Y.; Wilmarth, B.; Pagan, F.L.; et al. CSF microRNAs reveal impairment of angiogenesis and autophagy in Parkinson disease. Neurol. Genet. 2021, 7, e633. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Z.; Yang, X.; Li, T.; Liu, M.; Tang, H. miR-346 functions as a pro-survival factor under ER stress by activating mitophagy. Cancer Lett. 2018, 413, 69–81. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visintin, R.; Ray, S.K. Specific microRNAs for Modulation of Autophagy in Spinal Cord Injury. Brain Sci. 2022, 12, 247. https://doi.org/10.3390/brainsci12020247
Visintin R, Ray SK. Specific microRNAs for Modulation of Autophagy in Spinal Cord Injury. Brain Sciences. 2022; 12(2):247. https://doi.org/10.3390/brainsci12020247
Chicago/Turabian StyleVisintin, Rhett, and Swapan K. Ray. 2022. "Specific microRNAs for Modulation of Autophagy in Spinal Cord Injury" Brain Sciences 12, no. 2: 247. https://doi.org/10.3390/brainsci12020247
APA StyleVisintin, R., & Ray, S. K. (2022). Specific microRNAs for Modulation of Autophagy in Spinal Cord Injury. Brain Sciences, 12(2), 247. https://doi.org/10.3390/brainsci12020247