
A Metabolic Plasticity-Based Signature for Molecular Classification and Prognosis of Lower-Grade Glioma
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Glioma Datasets
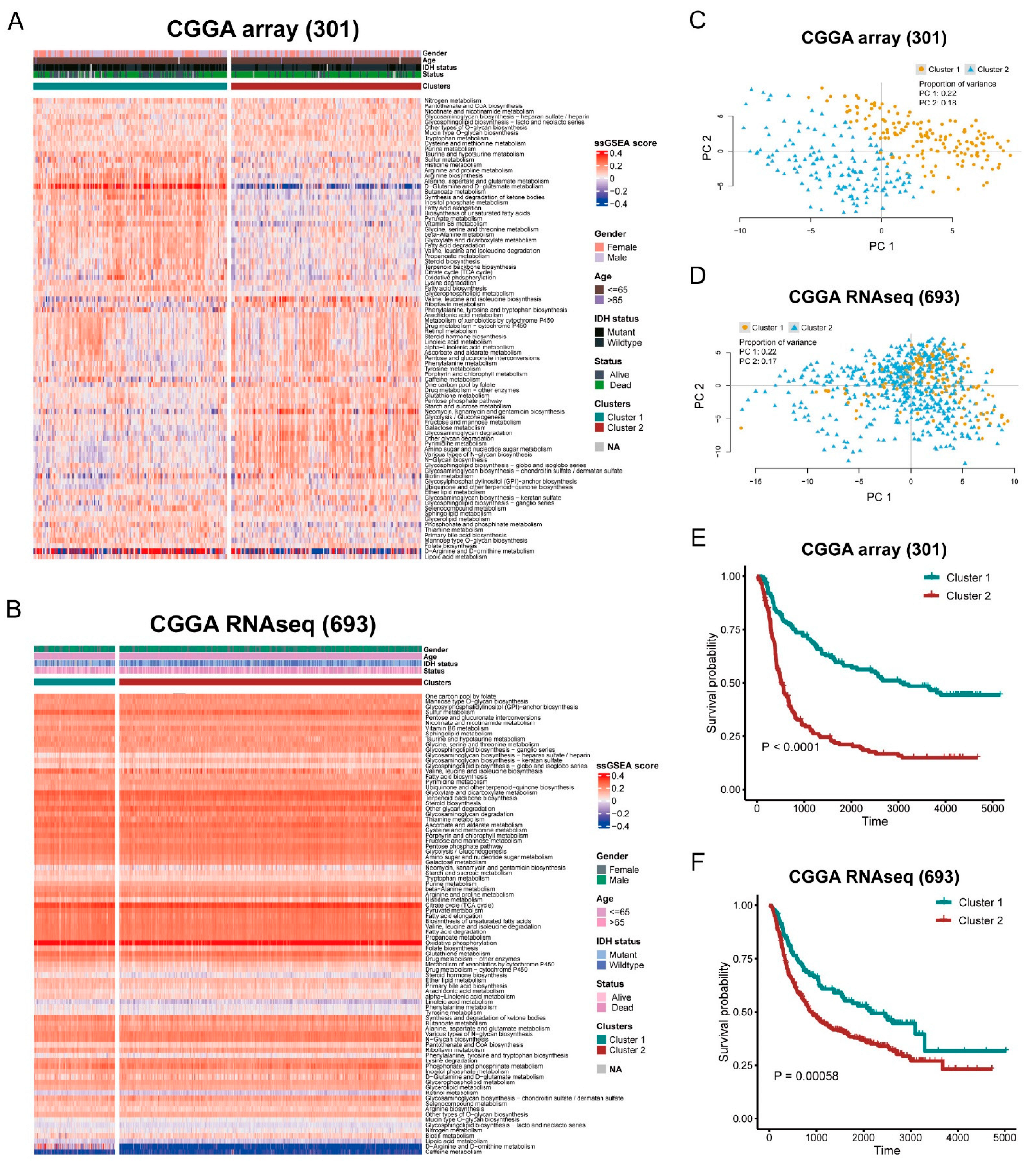
2.2. Metabolic Enrichment Based on Clustering
2.3. Comparison of Immune Infiltration between Clusters
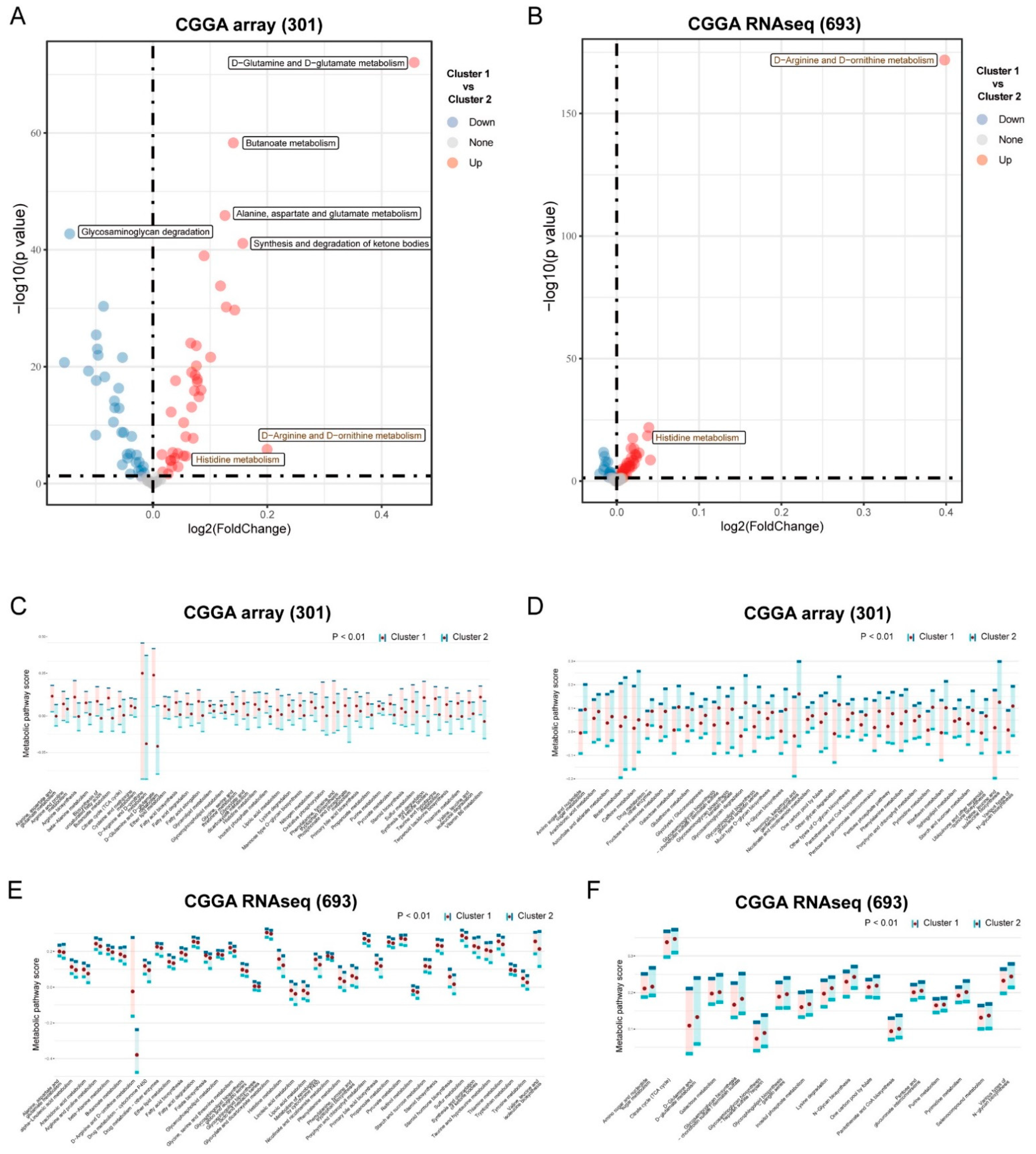
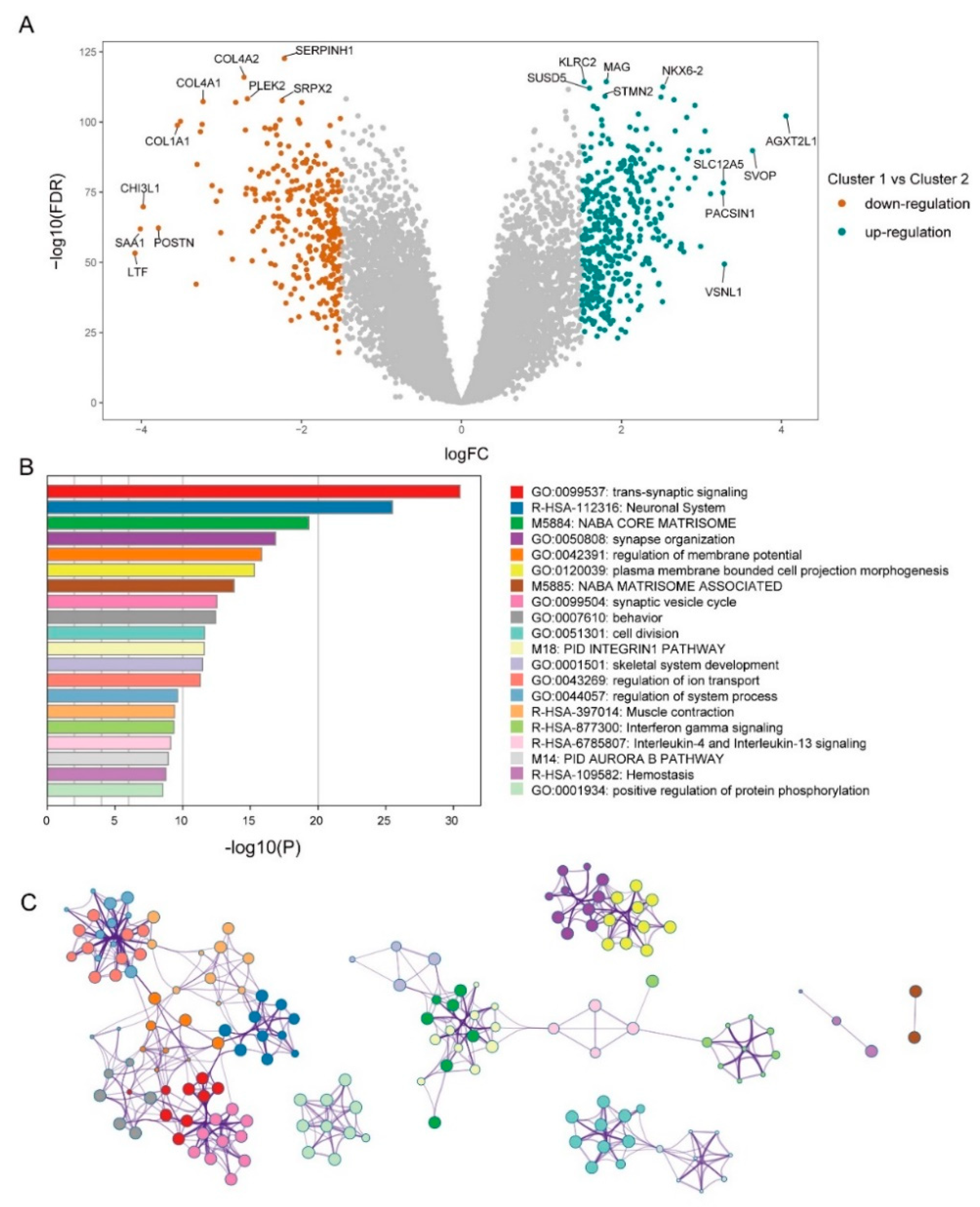
2.4. Analysis of Functional Differences between Clusters
2.5. Identification of Meaningful Co-Expression Module
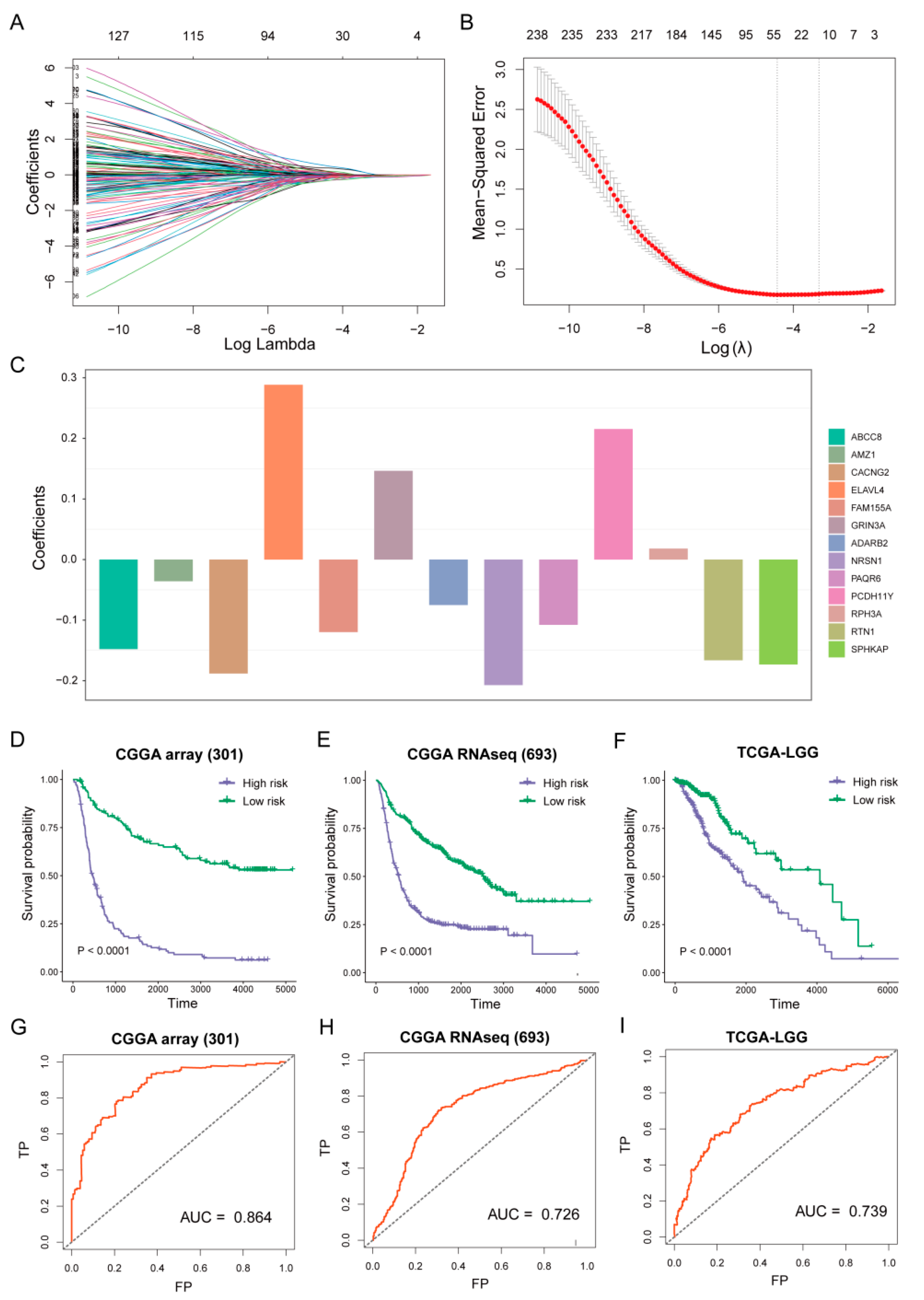
2.6. Prognostic Model Established
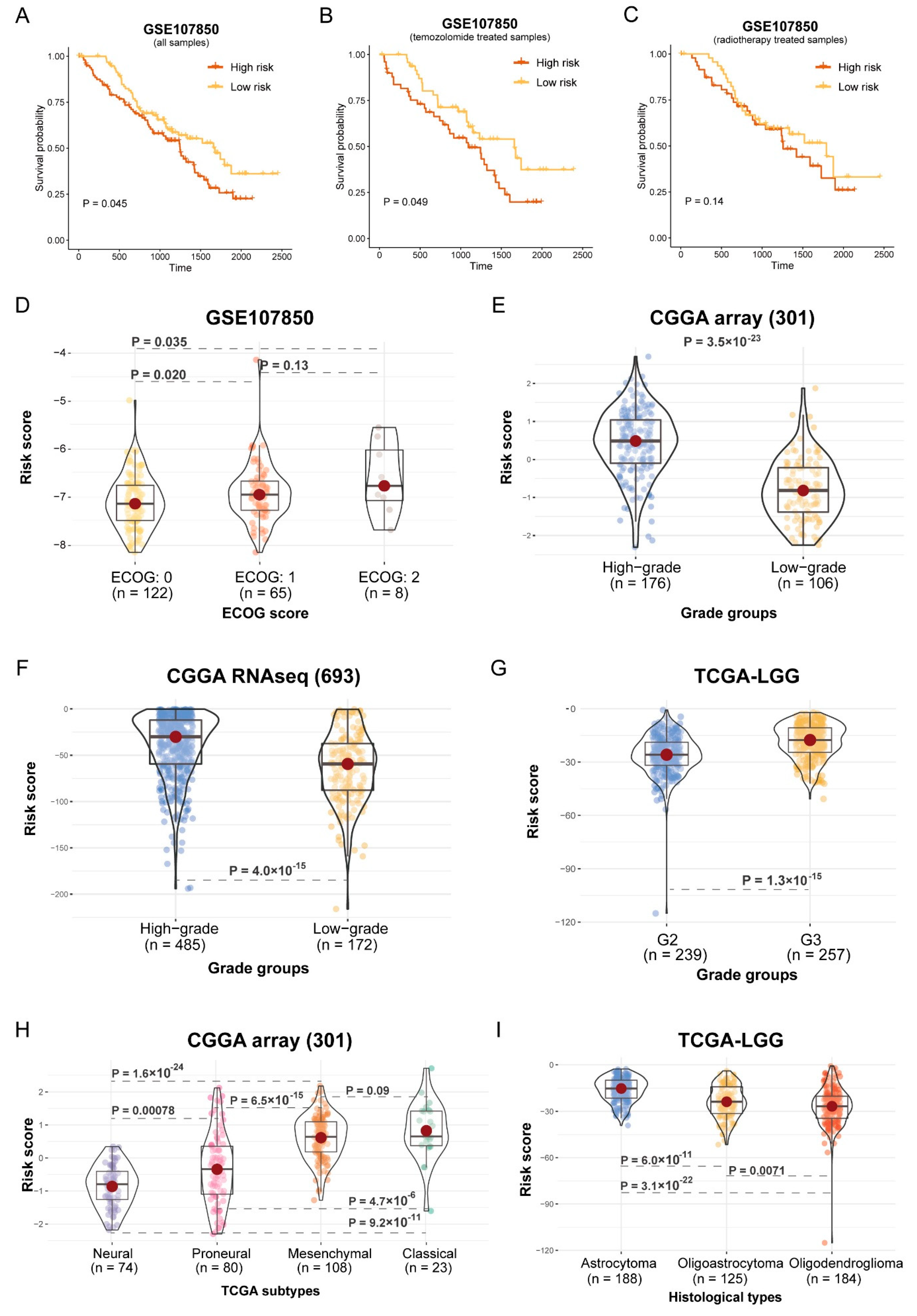
2.7. Survival Analysis and Correlation Analysis of Histological Subtypes and Risk Score
2.8. Quantitative Real-Time PCR and Cell Culture
2.9. Wound Healing and Transwell Migration Assay
2.10. Western Blotting
2.11. Cell Colony Formation Assay
2.12. Cell Proliferation Assay
3. Results
3.1. Stratification of Glioma Based on Metabolic Pathway
3.2. Differential Metabolism and Immune Infiltration between Clusters
3.3. Mining of Meaningful Module
3.4. Establish of Prognostic Model
3.5. Risk Score Associated with Histological Subtypes
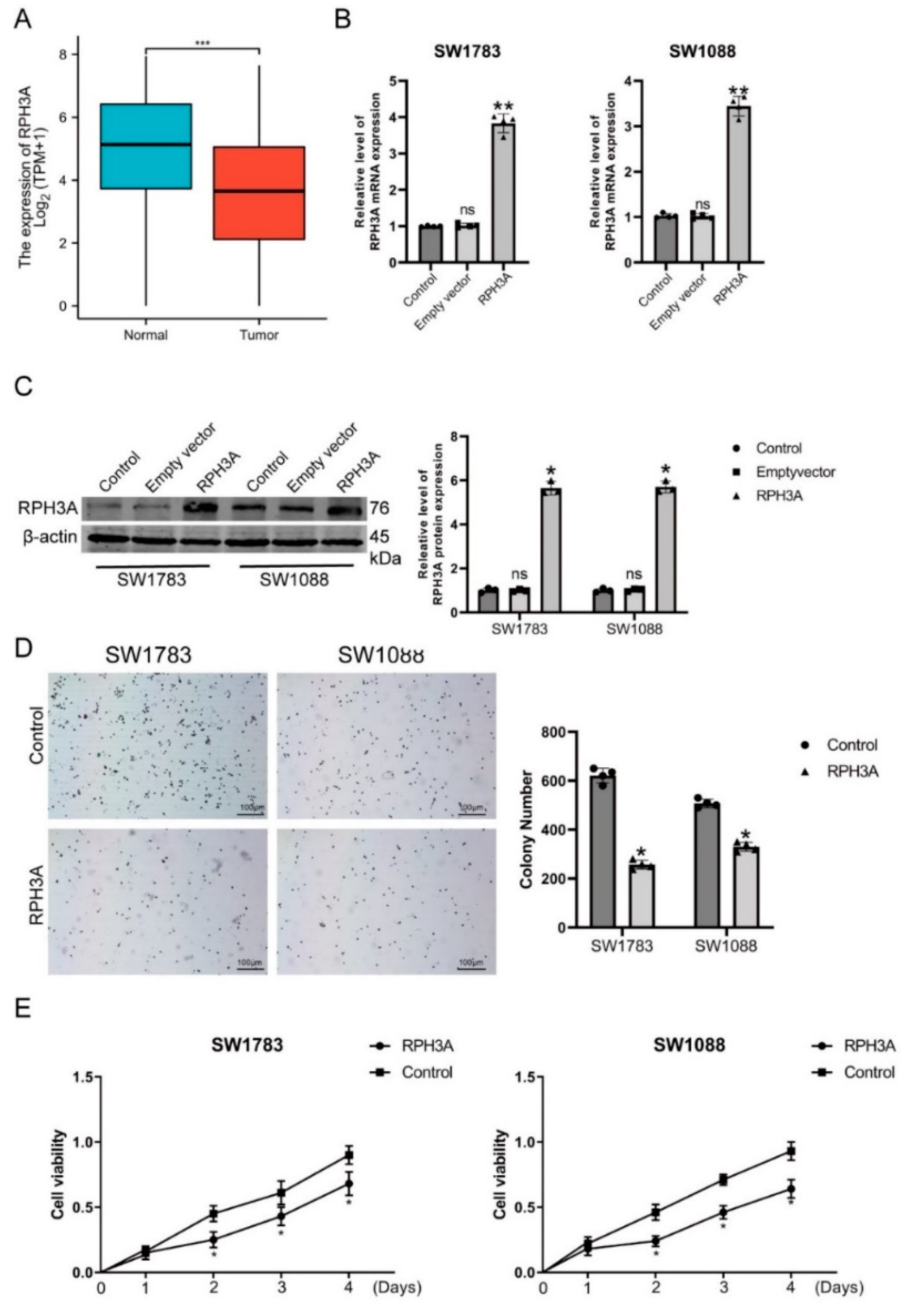
3.6. RPH3A Decreases LGG Cell Proliferation and Induce Apoptosis
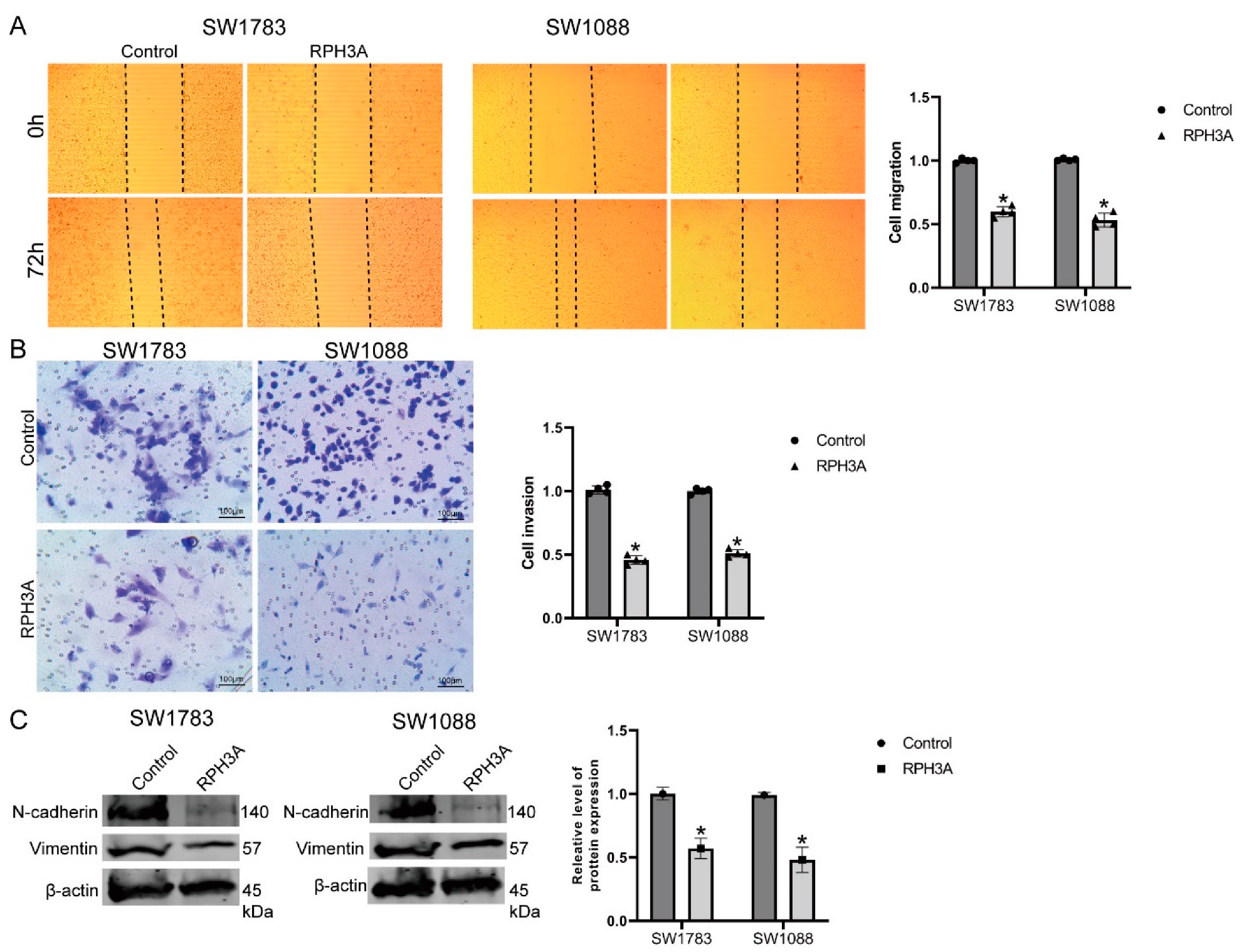
3.7. RPH3A Suppressed LGG Cell Migration, Invasion and EMT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.C.; Mehta, M.P. Low-Grade Glioma Radiotherapy Treatment and Trials. Neurosurg. Clin. North Am. 2019, 30, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.M.; Cahill, D.P.; Aldape, K.D.; Mehta, M.P. Treatment of Adult Lower-Grade Glioma in the Era of Genomic Medicine. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 75–81. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Shibao, S.; Minami, N.; Koike, N.; Fukui, N.; Yoshida, K.; Saya, H.; Sampetrean, O. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro Oncol. 2018, 20, 343–354. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef]
- Ghouzlani, A.; Kandoussi, S.; Tall, M.; Reddy, K.P.; Rafii, S.; Badou, A. Immune Checkpoint Inhibitors in Human Glioma Microenvironment. Front. Immunol. 2021, 12, 679425. [Google Scholar] [CrossRef]
- Yang, L.; Lin, P.C. Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin. Cancer Biol. 2017, 47, 185–195. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, X.; Chen, X.; Zhang, Q.; Hong, J. Novel Immune-Related Gene Signature for Risk Stratification and Prognosis of Survival in Lower-Grade Glioma. Front. Genet. 2020, 11, 363. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.Q.; Li, Y.T.; Yan, T.F.; Xu, Y.; Liu, B.H.; Yang, J.A.; Yang, X.; Chen, Q.X.; Zhang, H.B. Six Immune Associated Genes Construct Prognostic Model Evaluate Low-Grade Glioma. Front. Immunol. 2020, 11, 606164. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.; Zhong, Y.; Li, Q.; Li, Y.; Fan, H. Metabolic Remodeling in Glioma Immune Microenvironment: Intercellular Interactions Distinct from Peripheral Tumors. Front. Cell Dev. Biol. 2021, 9, 693215. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef]
- Rao, C.N.; Lakka, S.S.; Kin, Y.; Konduri, S.D.; Fuller, G.N.; Mohanam, S.; Rao, J.S. Expression of tissue factor pathway inhibitor 2 inversely correlates during the progression of human gliomas. Clin. Cancer Res. 2001, 7, 570–576. [Google Scholar]
- Kobayashi, M.; Mizutani, A.; Nishi, K.; Nakajima, S.; Shikano, N.; Nishii, R.; Fukuchi, K.; Kawai, K. Differences in accumulation and the transport mechanism of l- and d-methionine in high- and low-grade human glioma cells. Nucl. Med. Biol. 2017, 44, 78–82. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Chen, C.; Peng, H.; Lin, X.; Zhao, Q.; Chen, S.; Wang, X. Growth differentiation factor-15 overexpression promotes cell proliferation and predicts poor prognosis in cerebral lower-grade gliomas correlated with hypoxia and glycolysis signature. Life Sci. 2022, 302, 120645. [Google Scholar] [CrossRef]
- Zheng, Y.; Ji, Q.; Xie, L.; Wang, C.; Yu, C.N.; Wang, Y.L.; Jiang, J.; Chen, F.; Li, W.B. Ferroptosis-related gene signature as a prognostic marker for lower-grade gliomas. J. Cell Mol. Med. 2021, 25, 3080–3090. [Google Scholar] [CrossRef]
- Guo, J.C.; Wei, Q.S.; Dong, L.; Fang, S.S.; Li, F.; Zhao, Y. Prognostic Value of an Autophagy-Related Five-Gene Signature for Lower-Grade Glioma Patients. Front. Oncol. 2021, 11, 644443. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wu, Z.; Gu, J.; Zhu, Y.; Zhang, T.; Wang, F.; Huang, K.; Gu, C.; Xu, K.; Zhan, R.; et al. Metabolic Signature-Based Subtypes May Pave Novel Ways for Low-Grade Glioma Prognosis and Therapy. Front. Cell Dev. Biol. 2021, 9, 755776. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, D.; Lian, M. Identification of an epigenetic prognostic signature for patients with lower-grade gliomas. CNS Neurosci. Ther. 2021, 27, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Zhuang, S.M.; Xia, Z.; Xiao, Z.W.; Huang, C.X.; Su, Q.; Chen, J.; Liao, J. Pan-cancer analysis of GALNTs expression identifies a prognostic of GALNTs feature in low grade glioma. J. Leukoc. Biol. 2022, 741–5400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, S.; Guo, K.; Hu, Z.; Peng, J.; Li, J. Correlation and clinical significance of LC3, CD68+ microglia, CD4+ T lymphocytes, and CD8+ T lymphocytes in gliomas. Clin. Neurol. Neurosurg. 2018, 168, 167–174. [Google Scholar] [CrossRef]
- Qian, J.; Wang, C.; Wang, B.; Yang, J.; Wang, Y.; Luo, F.; Xu, J.; Zhao, C.; Liu, R.; Chu, Y. The IFN-gamma/PD-L1 axis between T cells and tumor microenvironment: Hints for glioma anti-PD-1/PD-L1 therapy. J. Neuroinflamm. 2018, 15, 290. [Google Scholar] [CrossRef]
- LaPorte, S.L.; Juo, Z.S.; Vaclavikova, J.; Colf, L.A.; Qi, X.; Heller, N.M.; Keegan, A.D.; Garcia, K.C. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell 2008, 132, 259–272. [Google Scholar] [CrossRef]
- Joshi, B.H.; Leland, P.; Lababidi, S.; Varrichio, F.; Puri, R.K. Interleukin-4 receptor alpha overexpression in human bladder cancer correlates with the pathological grade and stage of the disease. Cancer Med. 2014, 3, 1615–1628. [Google Scholar] [CrossRef]
- Todaro, M.; Lombardo, Y.; Francipane, M.G.; Alea, M.P.; Cammareri, P.; Iovino, F.; Di Stefano, A.B.; Di Bernardo, C.; Agrusa, A.; Condorelli, G.; et al. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ. 2008, 15, 762–772. [Google Scholar] [CrossRef]
- Setrerrahmane, S.; Xu, H. Tumor-related interleukins: Old validated targets for new anti-cancer drug development. Mol. Cancer 2017, 16, 153. [Google Scholar] [CrossRef]
- Scheurer, M.E.; Amirian, E.; Cao, Y.; Gilbert, M.R.; Aldape, K.D.; Kornguth, D.G.; El-Zein, R.; Bondy, M.L. Polymorphisms in the interleukin-4 receptor gene are associated with better survival in patients with glioblastoma. Clin. Cancer Res. 2008, 14, 6640–6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Yang, X.; Ji, C.; Tang, C.; Xie, L. Identification of an IL-4-Related Gene Risk Signature for Malignancy, Prognosis and Immune Phenotype Prediction in Glioma. Brain Sci. 2022, 12, 181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, J.; Han, C.; Liu, X.; Liu, J.; Yang, H. Identification of hub genes related to prognosis in glioma. Biosci. Rep. 2020, 40, BSR20193377. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Liu, Y.; Zhao, Z.; Wang, Y.; Huang, L.; Chai, R.; Li, G.; Jiang, T. ABCC8 mRNA expression is an independent prognostic factor for glioma and can predict chemosensitivity. Sci. Rep. 2020, 10, 12682. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Morshed, R.A.; Young, J.S.; Hervey-Jumper, S.L.; Berger, M.S. The management of low-grade gliomas in adults. J. Neurosurg. Sci. 2019, 63, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Yuan, Q.; Jian, X.; Randazzo, P.A.; Tang, W.; Wu, D. Small GTPase ARF6 Is a Coincidence-Detection Code for RPH3A Polarization in Neutrophil Polarization. J. Immunol. 2020, 204, 1012–1021. [Google Scholar] [CrossRef]
- Yuan, Q.; Ren, C.; Xu, W.; Petri, B.; Zhang, J.; Zhang, Y.; Kubes, P.; Wu, D.; Tang, W. PKN1 Directs Polarized RAB21 Vesicle Trafficking via RPH3A and Is Important for Neutrophil Adhesion and Ischemia-Reperfusion Injury. Cell Rep. 2017, 19, 2586–2597. [Google Scholar] [CrossRef]
- Zhu, X.; Li, H.; You, W.; Yu, Z.; Wang, Z.; Shen, H.; Li, X.; Yu, H.; Wang, Z.; Chen, G. Role of Rph3A in brain injury induced by experimental cerebral ischemia-reperfusion model in rats. CNS Neurosci. Ther. 2022, 28, 1124–1138. [Google Scholar] [CrossRef]
- Stanic, J.; Mellone, M.; Napolitano, F.; Racca, C.; Zianni, E.; Minocci, D.; Ghiglieri, V.; Thiolat, M.L.; Li, Q.; Longhi, A.; et al. Rabphilin 3A: A novel target for the treatment of levodopa-induced dyskinesias. Neurobiol. Dis. 2017, 108, 54–64. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.-C.; Wu, D.; Sun, H.; Wang, L.-K.; Chen, X.-F. A Metabolic Plasticity-Based Signature for Molecular Classification and Prognosis of Lower-Grade Glioma. Brain Sci. 2022, 12, 1138. https://doi.org/10.3390/brainsci12091138
Yang M-C, Wu D, Sun H, Wang L-K, Chen X-F. A Metabolic Plasticity-Based Signature for Molecular Classification and Prognosis of Lower-Grade Glioma. Brain Sciences. 2022; 12(9):1138. https://doi.org/10.3390/brainsci12091138
Chicago/Turabian StyleYang, Ming-Chun, Di Wu, Hui Sun, Lian-Kun Wang, and Xiao-Feng Chen. 2022. "A Metabolic Plasticity-Based Signature for Molecular Classification and Prognosis of Lower-Grade Glioma" Brain Sciences 12, no. 9: 1138. https://doi.org/10.3390/brainsci12091138