
Non-Association of Driver Alterations in PTEN with Differential Gene Expression and Gene Methylation in IDH1 Wildtype Glioblastomas


Abstract
:1. Introduction
2. Materials and Methods
2.1. Access to Data on Cbioportal
2.2. Primary Analyses of TCGA-FL-SG on GBs
2.2.1. Creation of Three Subsets of Cases Called Core Groups for Three Different Types of Analyses
2.2.2. Selection of Driver Genes for Analyses
2.2.3. Creating Groups with and without DAs in a Specific Gene
2.2.4. Associations between DAs in Individual Genes with DEMs, DMGs and DEPs
2.2.5. Associations between DAs in Individual Genes with Clinical Features and Survival
2.2.6. Association between PTEN and Other Analyzed Genes
2.2.7. Influence of Concurrent DAs in Other Drivers on the Non-Association of DAs in PTEN with DEMs and DMGs
2.2.8. Associations between Types of DA in PTEN with DEMs and DMGs
2.3. Secondary Analyses of Other Datasets Pertaining to DAs in PTEN
- (i).
- From the TCGA-FL-SG on GBs, using selection criteria such as those used to create the C1 group, a separate dataset was created using cases profiled on the U133 microarray platform (instead of the RNA sequencing v2 platform with RSEM, on which the primary analysis was performed). Groups of cases with and without DAs in PTEN were compared for DEMs.
- (ii).
- The IDH1-W-GBs in the Brain Tumor PDX (Mayo Clinic 2019) dataset were analyzed to determine the association of the DAs in PTEN with DEMs (in this dataset, the gene expression profile was available on the RNA sequencing v2 platform) and DMGs (in this dataset, the methylation test profile was available on the Illumina EPIC-850k platform) [45].
2.4. Additional Analyses
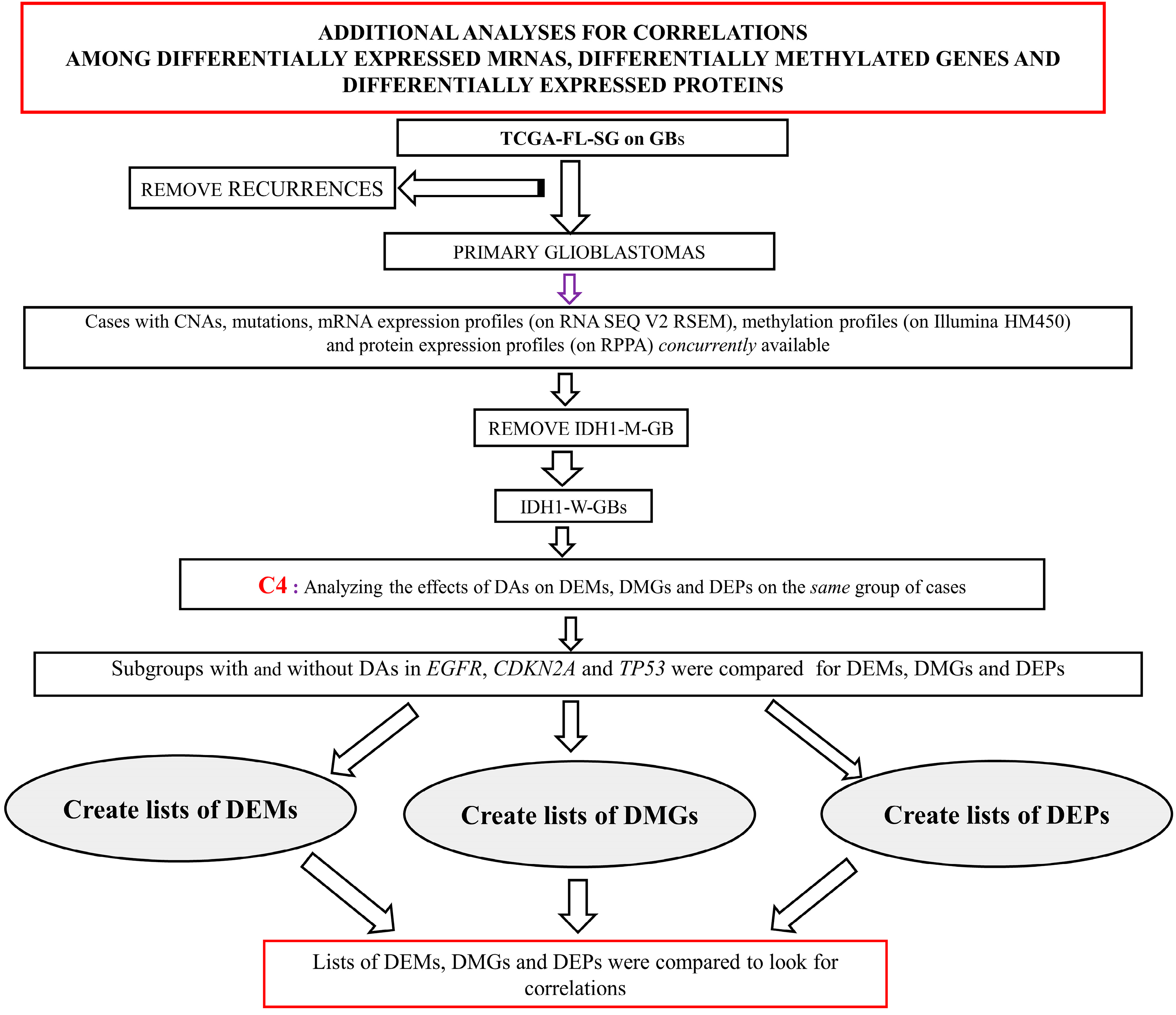
2.4.1. Analyses for Correlation among DEMs, DMGs and DEPs
2.4.2. Analyses for Enriched GO Terms and Pathways among DEMs and DMGs Associated with DAs in CDKN2A, EGFR and TP53
2.4.3. Analyses of DEMs and DMGs That Are Common between the DEMs and DMGs Associated with DAs in CDKN2A, EGFR and TP53
3. Results
3.1. Primary Analyses on the TCGA-FL-SG on GBs
3.1.1. Data Resources and Data Preparation
3.1.2. Associations between Individual DAs and DEMs
3.1.3. Associations between Individual DAs and DMGs
3.1.4. Associations between Individual DAs and DEPs
3.1.5. Statistical Analyses for Association between PTEN and other Genes
3.1.6. Associations between Individual DAs with Clinical Features and Survival
3.1.7. Influence of Concurrent DAs on the Non-Association of DAs in PTEN with DEMs and DMGs
3.1.8. Associations between Types of DAs in PTEN with DEMs and DMGs
3.2. Secondary Analyses on the Association between DAs in PTEN on Another Analysis Platform and Another Data Resource on GBs
3.3. Additional Analyses
3.3.1. Analyses for Correlation between DEMs, DMGs and DEPs
3.3.2. Analyses for Enriched GO Terms and Pathways among DEMs and DMGs Associated with DAs in CDKN2A, EGFR and TP53
3.3.3. Analyses of the DEMs and DMGs That Are Common between the DEMs and DMGs Associated with DAs in CDKN2A, EGFR and TP53
4. Discussion
4.1. The Fundamental Concepts That Guided the Basic Study Design
4.2. Selection of Datasets and Creation of User-Defined Groups
4.3. Associations of DAs in PTEN with Differential mRNA Expressions and Gene Methylations
4.4. Associations between DAs and Differential Protein Expression
4.5. Associations between DAs with Clinical Features and Survival Figures
4.6. Associations among DEMs, DMGs and DEPs in a Single Group of Cases
4.7. Determining the Biological Relevance of DEMs and DMGs Associated with DAs in CDKN2A, EGFR and TP53
4.8. Analyses of the DEMs and DMGs That Are Common between the DEMs and DMGs Associated with DAs in CDKN2A, EGFR and TP53
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PTEN | Phosphatase and Tensin homolog |
| DA | Driver alteration |
| CAN | Copy number alteration |
| DEM | Differentially expressed mRNA |
| DMG | Differentially methylated gene |
| DEP | Differentially expressed protein |
| TCGA-FL-SG | The Cancer Genome Atlas Firehose Legacy Study Group |
| GB | Glioblastoma |
| IDH1-W-GB | IDH1 Wildtype Glioblastoma |
| IDH1-M-GB | IDH1 Mutant Glioblastoma |
| UREG | Upregulated |
| DREG | Downregulated |
| HYPERMETH | Hypermethylated |
| HYPOMETH | Hypomethylated |
| GENE ONTOLOGY | Gene ontology |
References
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Wodarz, D.; Newell, A.C.; Komarova, N.L. Passenger mutations can accelerate tumour suppressor gene inactivation in cancer evolution. J. R. Soc. Interface 2018, 15, 20170967. [Google Scholar] [CrossRef] [Green Version]
- Korenjak, M.; Zavadil, J. Experimental identification of cancer driver alterations in the era of pan-cancer genomics. Cancer Sci. 2019, 110, 3622–3629. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.L. A Novel Method for Identifying the Potential Cancer Driver Genes Based on Molecular Data Integration. Biochem. Genet. 2020, 58, 16–39. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Loeb, L.A. Mutator phenotype in cancer: Origin and consequences. Semin. Cancer Biol. 2010, 20, 279–280. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.A.; Cho, D.Y.; Przytycka, T.M. Understanding Genotype-Phenotype Effects in Cancer via Network Approaches. PLoS Comput. Biol 2016, 12, e1004747. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic significance of epidermal growth factor receptor expression in glioma patients. OncoTargets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.Y.; Ryu, J.S.; Sim, Y.S.; Kim, D.; Lee, S.Y.; Choi, J.; Park, S.; Ryu, Y.J.; Lee, J.H.; Chang, J.H. Clinical significance of EGFR mutation types in lung adenocarcinoma: A multi-centre Korean study. PLoS ONE 2020, 15, e0228925. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Valencia, A.; Godzik, A. Understanding oncogenicity of cancer driver genes and mutations in the cancer genomics era. FEBS Lett. 2020, 594, 4233–4246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; He, D.N.; Wu, Z.Y.; Zhu, X.; Wen, X.L.; Li, X.H.; Guo, Y.; Wang, H.J.; Wang, Z.Z. Oncogenic signaling pathway dysregulation landscape reveals the role of pathways at multiple omics levels in pan-cancer. Front. Genet. 2022, 13, 916400. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Peng, W.; Wang, F.; Wang, J. Identifying driver genes involving gene dysregulated expression, tissue-specific expression and gene-gene network. BMC Med. Genom. 2019, 12, 168. [Google Scholar] [CrossRef] [Green Version]
- Jia, P.; Zhao, Z. Impacts of somatic mutations on gene expression: An association perspective. Brief. Bioinform. 2017, 18, 413–425. [Google Scholar] [CrossRef]
- Landan, G.; Cohen, N.M.; Mukamel, Z.; Bar, A.; Molchadsky, A.; Brosh, R.; Horn-Saban, S.; Zalcenstein, D.A.; Goldfinger, N.; Zundelevich, A.; et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet. 2012, 44, 1207–1214. [Google Scholar] [CrossRef]
- Lee, C.J.; Ahn, H.; Jeong, D.; Pak, M.; Moon, J.H.; Kim, S. Impact of mutations in DNA methylation modification genes on genome-wide methylation landscapes and downstream gene activations in pan-cancer. BMC Med. Genom. 2020, 13, 27. [Google Scholar] [CrossRef]
- Mehnert, M.; Ciuffa, R.; Frommelt, F.; Uliana, F.; van Drogen, A.; Ruminski, K.; Gstaiger, M.; Aebersold, R. Multi-layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes. Nat. Commun. 2020, 11, 3563. [Google Scholar] [CrossRef]
- Halvey, P.J.; Zhang, B.; Coffey, R.J.; Liebler, D.C.; Slebos, R.J. Proteomic consequences of a single gene mutation in a colorectal cancer model. J. Proteome Res. 2012, 11, 1184–1195. [Google Scholar] [CrossRef]
- Pettini, F.; Visibelli, A.; Cicaloni, V.; Iovinelli, D.; Spiga, O. Multi-Omics Model Applied to Cancer Genetics. Int. J. Mol. Sci. 2021, 22, 5751. [Google Scholar] [CrossRef] [PubMed]
- Al-Thoubaity, F.K. Molecular classification of breast cancer: A retrospective cohort study. Ann. Med. Surg. 2020, 49, 44–48. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Chai, S.; Chen, W.; Yu, B. DNA methylation profiling identifies potentially significant epigenetically-regulated genes in glioblastoma multiforme. Oncol. Lett. 2019, 18, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, J.; Dai, X. DNA methylation profiles capturing breast cancer heterogeneity. BMC Genom. 2019, 20, 823. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Cano, S.J. Tumor heterogeneity: Mechanisms and bases for a reliable application of molecular marker design. Int. J. Mol. Sci. 2012, 13, 1951–2011. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, S.; Pan, L. Identification of key differentially expressed mRNAs and microRNAs in non-small cell lung cancer using bioinformatics analysis. Exp. Ther. Med. 2020, 20, 3720–3732. [Google Scholar] [CrossRef]
- Wang, A.; Zhang, G. Differential gene expression analysis in glioblastoma cells and normal human brain cells based on GEO database. Oncol. Lett. 2017, 14, 6040–6044. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Rudland, P.S.; Sibson, D.R.; Barraclough, R. Identification of mRNAs differentially-expressed between benign and malignant breast tumour cells. Br. J. Cancer 2002, 87, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Xu, M.; Wang, L.; Zhou, W.; Xiang, R.; Shi, Y.; Zhang, Y.; Piao, Y. Integrative analysis of DNA methylation and gene expression identified cervical cancer-specific diagnostic biomarkers. Signal Transduct. Target. Ther. 2019, 4, 55. [Google Scholar] [CrossRef] [Green Version]
- Vaissiere, T.; Hung, R.J.; Zaridze, D.; Moukeria, A.; Cuenin, C.; Fasolo, V.; Ferro, G.; Paliwal, A.; Hainaut, P.; Brennan, P.; et al. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009, 69, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraboth, Z.; Galik, B.; Tompa, M.; Kajtar, B.; Urban, P.; Gyenesei, A.; Miseta, A.; Kalman, B. DNA CpG methylation in sequential glioblastoma specimens. J. Cancer Res. Clin. Oncol. 2020, 146, 2885–2896. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lih, T.M.; Pan, J.; Hoti, N.; Dong, M.; Cao, L.; Hu, Y.; Cho, K.C.; Chen, S.Y.; Eguez, R.V.; et al. Proteomic signatures of 16 major types of human cancer reveal universal and cancer-type-specific proteins for the identification of potential therapeutic targets. J. Hematol. Oncol. 2020, 13, 170. [Google Scholar] [CrossRef]
- Suk, K. Proteomic analysis of glioma chemoresistance. Curr. Neuropharmacol. 2012, 10, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Wang, S.; Zhang, Y.H.; Cai, Y.D.; Liu, H. Analysis of Important Gene Ontology Terms and Biological Pathways Related to Pancreatic Cancer. BioMed Res. Int. 2016, 2016, 7861274. [Google Scholar] [CrossRef] [Green Version]
- Paczkowska, M.; Barenboim, J.; Sintupisut, N.; Fox, N.S.; Zhu, H.; Abd-Rabbo, D.; Mee, M.W.; Boutros, P.C.; Drivers, P.; Functional Interpretation Working, G.; et al. Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 2020, 11, 735. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Garbe, M.; Friedrich, B.; Mittelbronn, M.; Klink, B. Comparative transcriptomics reveals similarities and differences between astrocytoma grades. BMC Cancer 2015, 15, 952. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.H.; Zhang, C.; Berry, G.J.; Altman, R.B.; Re, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 12474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.T.; Lee, Y.M.; Huang, R.S. The impact of the Cancer Genome Atlas on lung cancer. Transl. Res. 2015, 166, 568–585. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Karim, M.A.; Samad, A.; Adhikari, U.K.; Kader, M.A.; Kabir, M.M.; Islam, M.A.; Hasan, M.N. A Multi-Omics Analysis of Bone Morphogenetic Protein 5 (BMP5) mRNA Expression and Clinical Prognostic Outcomes in Different Cancers Using Bioinformatics Approaches. Biomedicines 2020, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Li, H.; Yan, Y.; Zang, Y.; Li, K.; Wang, Y.; Xue, F. Identification of key genes and pathways between type I and type II endometrial cancer using bioinformatics analysis. Oncol. Lett. 2019, 18, 2464–2476. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Jing, X.; Yi, Q.; Long, C.; Tan, B.; Li, X.; Chen, X.; Huang, Y.; Xiang, Z.; Tian, J.; et al. Systematic analysis of gene expression alterations and clinical outcomes of STAT3 in cancer. Oncotarget 2018, 9, 3198–3213. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Meng, Y. SUMOylation Regulator-Related Molecules Can Be Used as Prognostic Biomarkers for Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 658856. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA A Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E. Friend or foe-IDH1 mutations in glioma 10 years on. Carcinogenesis 2019, 40, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.I.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar]
- Benitez, J.A.; Ma, J.; D’Antonio, M.; Boyer, A.; Camargo, M.F.; Zanca, C.; Kelly, S.; Khodadadi-Jamayran, A.; Jameson, N.M.; Andersen, M.; et al. PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nat. Commun. 2017, 8, 15223. [Google Scholar] [CrossRef] [Green Version]
- Hill, V.K.; Kim, J.S.; James, C.D.; Waldman, T. Correction of PTEN mutations in glioblastoma cell lines via AAV-mediated gene editing. PLoS ONE 2017, 12, e0176683. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Gotea, V.; Margolin, G.; Elnitski, L. Significant associations between driver gene mutations and DNA methylation alterations across many cancer types. PLoS Comput. Biol. 2017, 13, e1005840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Liguori, M.J. Comparison of RNA-Seq and Microarray Gene Expression Platforms for the Toxicogenomic Evaluation of Liver From Short-Term Rat Toxicity Studies. Front. Genet. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, B.D. A Practical Guide to the Measurement and Analysis of DNA Methylation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.D.; Hodakoski, C.; Barrows, D.; Mense, S.M.; Parsons, R.E. PTEN function: The long and the short of it. Trends Biochem. Sci. 2014, 39, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Li, S.; Wang, F.; Fan, C.; Wang, J. Identification of key pathways and genes in PTEN mutation prostate cancer by bioinformatics analysis. BMC Med. Genet. 2019, 20, 191. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Lehner, B. Cancer type-dependent genetic interactions between cancer driver alterations indicate plasticity of epistasis across cell types. Mol. Syst. Biol. 2015, 11, 824. [Google Scholar] [CrossRef]
- Temko, D.; Tomlinson, I.P.M.; Severini, S.; Schuster-Bockler, B.; Graham, T.A. The effects of mutational processes and selection on driver mutations across cancer types. Nat. Commun. 2018, 9, 1857. [Google Scholar] [CrossRef] [Green Version]
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.; Andrieux, G.; Ahmed, M.; Chakraborty, S. Integration of Online Omics-Data Resources for Cancer Research. Front. Genet. 2020, 11, 578345. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Cui, H. The Emerging Roles of RNA Modifications in Glioblastoma. Cancers 2020, 12, 736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rynkeviciene, R.; Simiene, J.; Strainiene, E.; Stankevicius, V.; Usinskiene, J.; Miseikyte Kaubriene, E.; Meskinyte, I.; Cicenas, J.; Suziedelis, K. Non-Coding RNAs in Glioma. Cancers 2018, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Raman, A.T.; Wang, X.; Gaiti, F.; Chaligne, R.; Mohammad, A.W.; Arczewska, A.; Smith, Z.D.; Landau, D.A.; Aryee, M.J.; et al. Smart-RRBS for single-cell methylome and transcriptome analysis. Nat. Protoc. 2021, 16, 4004–4030. [Google Scholar] [CrossRef] [PubMed]
- Nicora, G.; Vitali, F.; Dagliati, A.; Geifman, N.; Bellazzi, R. Integrated Multi-Omics Analyses in Oncology: A Review of Machine Learning Methods and Tools. Front. Oncol. 2020, 10, 1030. [Google Scholar] [CrossRef]
- Mallik, M.K. An attempt to understand glioma stem cell biology through centrality analysis of a protein interaction network. J. Theor. Biol. 2018, 438, 78–91. [Google Scholar] [CrossRef]
- Stanta, G.; Bonin, S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front. Med. 2018, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Muscat, A.M.; Wong, N.C.; Drummond, K.J.; Algar, E.M.; Khasraw, M.; Verhaak, R.; Field, K.; Rosenthal, M.A.; Ashley, D.M. The evolutionary pattern of mutations in glioblastoma reveals therapy-mediated selection. Oncotarget 2018, 9, 7844–7858. [Google Scholar] [CrossRef] [Green Version]
- Giotta Lucifero, A.; Luzzi, S. Immune Landscape in PTEN-Related Glioma Microenvironment: A Bioinformatic Analysis. Brain Sci. 2022, 12, 501. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015, 5, 10775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, F.; Hu, R.; Yang, H.; Liu, J.; Sui, J.; Xiang, X.; Wang, F.; Chu, L.; Song, S. PTEN gene mutations correlate to poor prognosis in glioma patients: A meta-analysis. OncoTargets Ther. 2016, 9, 3485–3492. [Google Scholar] [CrossRef] [Green Version]
- Koike, N.; Kassai, Y.; Kouta, Y.; Miwa, H.; Konishi, M.; Itoh, N. Brorin, a novel secreted bone morphogenetic protein antagonist, promotes neurogenesis in mouse neural precursor cells. J. Biol. Chem. 2007, 282, 15843–15850. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, S.D.; Zhou, S.; Huse, J.T.; de Groot, J.F.; Xiu, J.; Subramaniam, D.S.; Mehta, S.; Gatalica, Z.; Swensen, J.; Sanai, N.; et al. Targetable Gene Fusions Associate With the IDH Wild-Type Astrocytic Lineage in Adult Gliomas. J. Neuropathol. Exp. Neurol. 2018, 77, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Pan, Y.; Yan, M.; Bao, G.; Sun, X. Identification of potential crucial genes and molecular mechanisms in glioblastoma multiforme by bioinformatics analysis. Mol. Med. Rep. 2020, 22, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, Q. Identification of Core Genes and Screening of Potential Targets in Glioblastoma Multiforme by Integrated Bioinformatic Analysis. Front. Oncol. 2020, 10, 615976. [Google Scholar] [CrossRef]
- Gubanova, N.V.; Orlova, N.G.; Dergilev, A.I.; Oparina, N.Y.; Orlov, Y.L. Glioblastoma gene network reconstruction and ontology analysis by online bioinformatics tools. J. Integr. Bioinform. 2021, 18, 20210031. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Concurrent DAs in Other Driver Genes * along with DAs in PTEN | Groups Compared to Determine the Influence of the Presence or Absence of Other Concurrent DAs on the Non-Association of DAs in PTEN with DEMs and DMGs |
|---|---|
| DAs in CDKN2A present | CDKN2A + PTEN + vs. CDKN2A + PTEN- |
| DAs in CDKN2A absent | CDKN2A—PTEN + vs. CDKN2A—PTEN- |
| DAs in EGFR present | EGFR + PTEN + vs. EGFR + PTEN- |
| DAs in EGFR absent | EGFR—PTEN + vs. EGFR—PTEN- |
| DAs in TP53 present | TP53 + PTEN + vs. TP53 + PTEN- |
| DAs in TP53 absent | TP53—PTEN + vs. TP53—PTEN- |
| DAs in RB1 present | RB1 + PTEN + vs. RB1 + PTEN- |
| DAs in RB1 are absent | RB1—PTEN + vs. RB1—PTEN- |
| Genes with DAs | Number of Cases with DAs and the Corresponding Prevalence in Group C1 | Number of Upregulated mRNAs in Group with DAs Compared to Group without DAs | Number of Downregulated mRNAs in Group with DAs Compared to Group without DAs | Total of Differentially Expressed mRNAs |
|---|---|---|---|---|
| CDKN2A | 75 (58.13%) | 81 | 221 | 302 |
| EGFR | 75 (58.13%) | 613 | 664 | 1277 |
| PTEN | 52 (40.31%) | 0 | 0 | 0 |
| TP53 | 38 (29.45%) | 1000 | 285 | 1285 |
| CDK4 | 23 (17.83%) | 794 | 722 | 1516 |
| PDGFRA | 20 (15.50%) | 28 | 4 | 32 |
| MDM4 | 12 (9.30%) | 11 | 7 | 18 |
| RB1 | 14 (10.85%) | 94 | 76 | 170 |
| PIK3CA | 9 (6.97%) | 4 | 9 | 13 |
| NF1 | 12 (9.30%) | 85 | 152 | 237 |
| MDM2 | 9 (6.97%) | 124 | 176 | 300 |
| PIK3R1 | 6 (4.65%) | 19 | 18 | 37 |
| Genes with DAs | Number of Cases with DAs and the Corresponding Prevalence in Group C2 | Number of Hypermethylated Genes in Group with DAs Compared to Group without DAs | Number of Hypomethylated Genes in Group with DAs Compared to Group without DAs | Total of Differentially Methylated Genes |
|---|---|---|---|---|
| CDKN2A | 67 (65.68%) | 1 | 2 | 3 |
| EGFR | 44 (43.13%) | 34 | 9 | 43 |
| PTEN | 32 (31.37%) | 0 | 0 | 0 |
| TP53 | 22 (21.56%) | 2 | 22 | 24 |
| CDK4 | 16 (15.68%) | 9 | 3 | 12 |
| PDGFRA | 14 (13.72%) | 1 | 18 | 19 |
| MDM4 | 9 (8.82%) | 0 | 53 | 53 |
| RB1 | 8 (7.84%) | 208 | 61 | 269 |
| PIK3CA | 12 (11.76%) | 0 | 7 | 7 |
| NF1 | 16 (15.68%) | 639 | 135 | 774 |
| MDM2 | 11 (10.78%) | 10 | 0 | 10 |
| PIK3R1 | 15 (14.70%) | 0 | 0 | 0 |
| Genes with DAs | Number of Cases with DAs and the Corresponding Prevalence in Group C3 | Number of Overexpressed Proteins in Group with DAs Compared to Group without DAs | Number of Underexpressed Proteins in Group with DAs Compared to Group without DAs | Total Differentially Expressed Proteins |
|---|---|---|---|---|
| CDKN2A | 89 (67.42%) | 1 | 0 | 1 |
| EGFR | 71 (53.78%) | 8 | 6 | 14 |
| PTEN | 44 (33.33%) | 6 | 0 | 6 |
| TP53 | 37 (28.03%) | 0 | 3 | 3 |
| CDK4 | 22 (16.67%) | 0 | 0 | 0 |
| PDGFRA | 19 (14.39%) | 0 | 0 | 0 |
| PIK3R1 | 15 (11.36%) | 0 | 0 | 0 |
| PIK3CA | 11 (8.33%) | 0 | 2 | 2 |
| MDM2 | 11 (8.33%) | 0 | 0 | 0 |
| MDM4 | 13 (9.84%) | 0 | 0 | 0 |
| NF1 | 17 (12.87%) | 1 | 4 | 5 |
| RB1 | 12 (9.09%) | 0 | 0 | 0 |
| Groups Compared | Upregulated mRNA Count | Downregulated mRNA Count | Hypermethylated Gene Count | Hypomethylated Gene Count |
|---|---|---|---|---|
| CDKN2A+PTEN+ (29)/[19] vs. CDKN2A+PTEN- (46)/[48] | 0 | 0 | 0 | 0 |
| CDKN2A-PTEN+ (23)/[13] vs. CDKN2A-PTEN- (31)/[22] | 0 | 0 | 0 | 0 |
| EGFR+PTEN+ (28)/[13] vs. EGFR+PTEN- (47)/[31] | 0 | 0 | 0 | 0 |
| EGFR-PTEN+ (24)/[19] vs. EGFR-PTEN- (30)/[39] | 0 | 0 | 0 | 0 |
| TP53+PTEN+ (16)/[7] vs. TP53+PTEN- (22)/[15] | 0 | 0 | 0 | 0 |
| TP53-PTEN+ (36)/[25] vs. TP53-PTEN- (55)/[55] | 0 | 0 | 0 | 0 |
| RB1+PTEN+ (10)/[4] vs. RB1+PTEN- (4)/[4] | 0 | 1 | 0 | 0 |
| RB1-PTEN+ (42)/[28] vs. RB1- PTEN- (73)/[62] | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallik, M.K.; Majumdar, K.; Mujtaba, S. Non-Association of Driver Alterations in PTEN with Differential Gene Expression and Gene Methylation in IDH1 Wildtype Glioblastomas. Brain Sci. 2023, 13, 186. https://doi.org/10.3390/brainsci13020186
Mallik MK, Majumdar K, Mujtaba S. Non-Association of Driver Alterations in PTEN with Differential Gene Expression and Gene Methylation in IDH1 Wildtype Glioblastomas. Brain Sciences. 2023; 13(2):186. https://doi.org/10.3390/brainsci13020186
Chicago/Turabian StyleMallik, Mrinmay Kumar, Kaushik Majumdar, and Shiraz Mujtaba. 2023. "Non-Association of Driver Alterations in PTEN with Differential Gene Expression and Gene Methylation in IDH1 Wildtype Glioblastomas" Brain Sciences 13, no. 2: 186. https://doi.org/10.3390/brainsci13020186