
The Genetics of Intellectual Disability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Phenotype-First Approach
3. Genotype-First Approach
4. The Era of Next-Generation Sequencing
5. Gene Discovery in ID
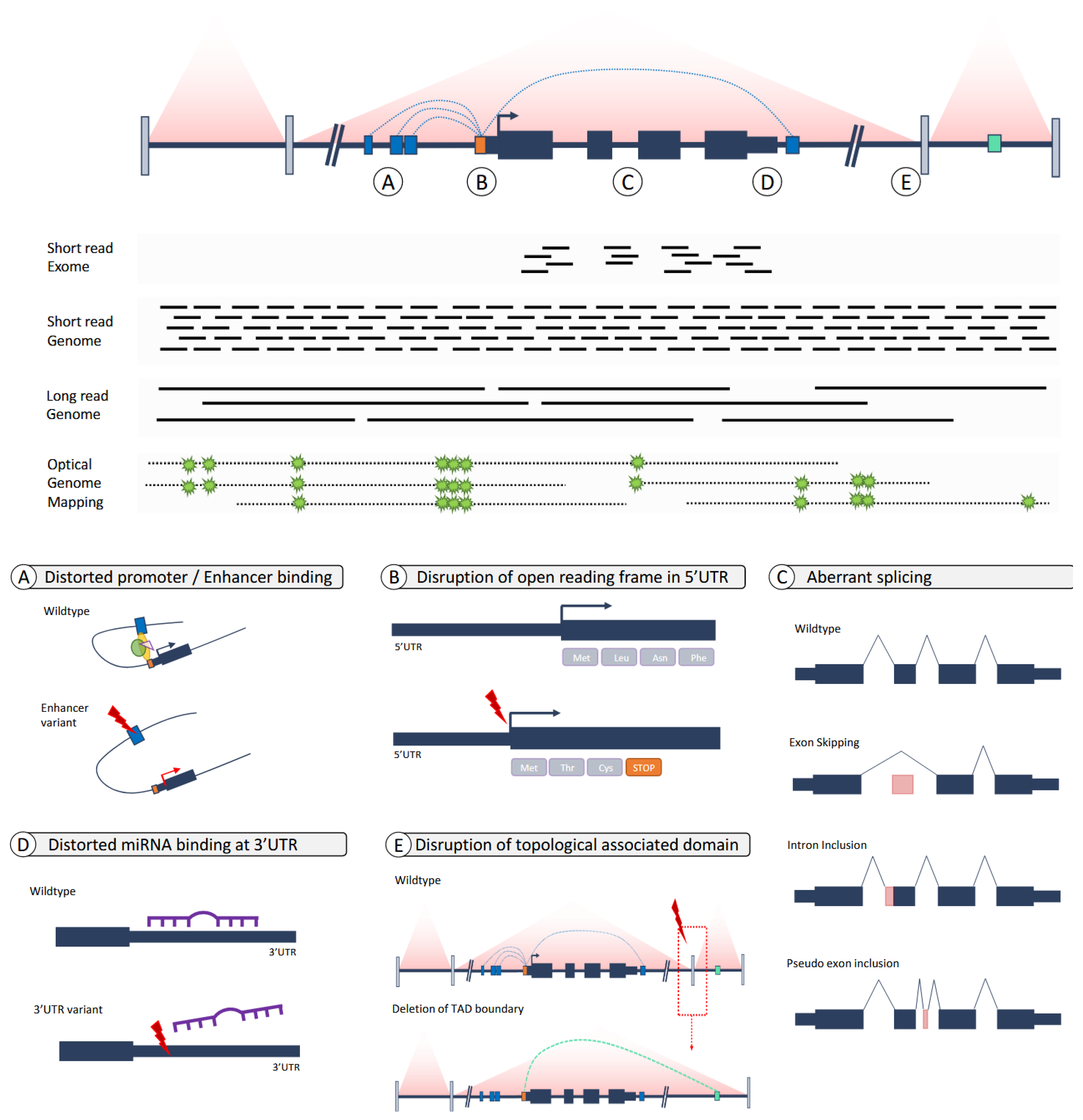
6. From Exomes to Genomes—From Coding to Non-Coding Variants
7. Genomes: From Short Reads to Long Reads and The Incorporation of Other -Omics
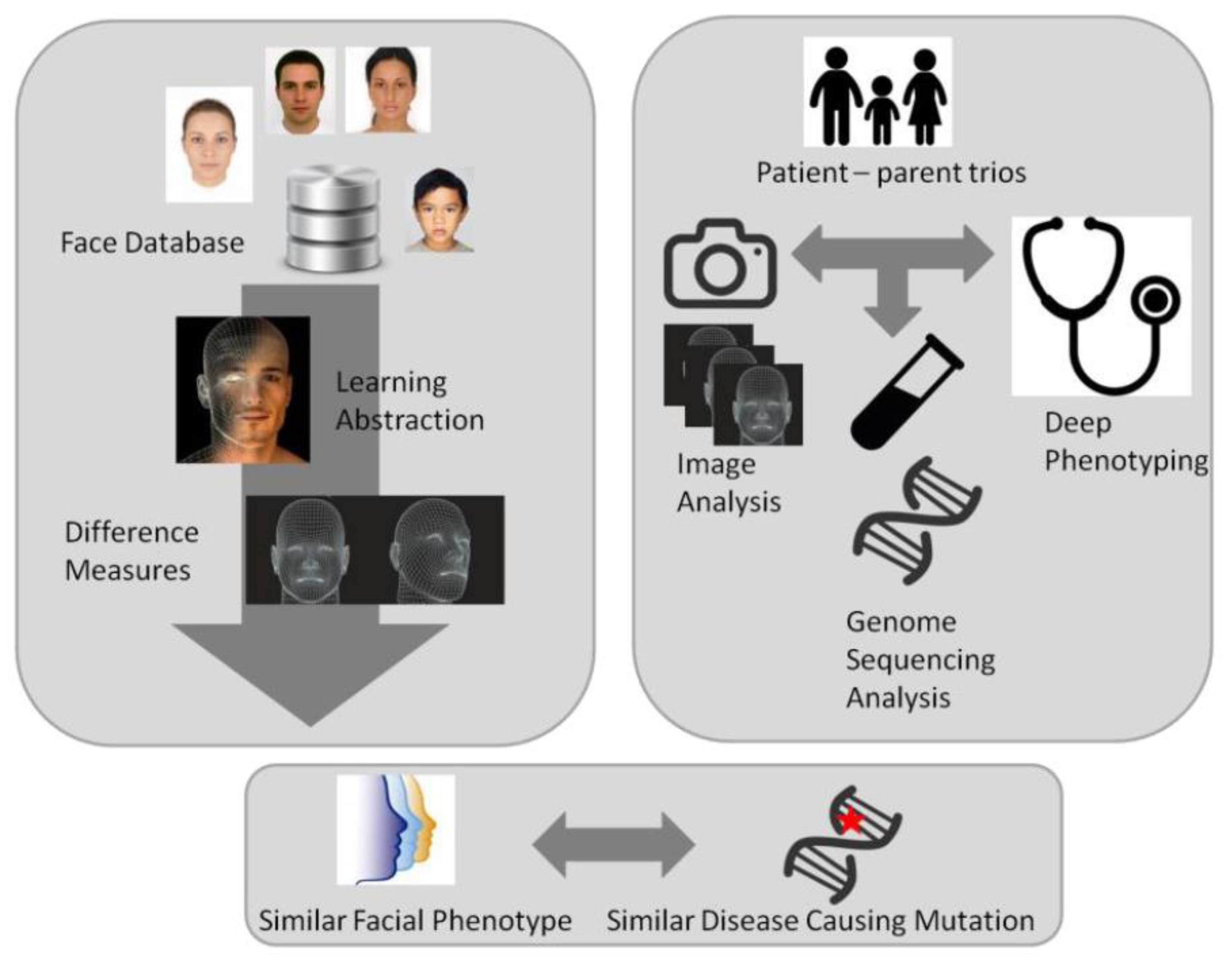
8. Quantitative Phenotyping
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association, DSM V. 2013. Available online: http://www.dsm5.org (accessed on 1 November 2022).
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D. Practice parameter: Evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, J.; Zitman, F.G. It is time to bring borderline intellectual functioning back into the main fold of classification systems. BJPsych Bull. 2016, 40, 204–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maulik, P.K.; Mascarenhas, M.N.; Mathers, C.D.; Dua, T.; Saxena, S. Prevalence of intellectual disability: A meta-analysis of population-based studies. Res. Dev. Disabil. 2011, 32, 419–436. [Google Scholar] [CrossRef]
- Spindler, U.P.; Hotopp, L.C.; Bach, V.A.; Hornemann, F.; Syrbe, S.; Andreas, A.; Merkenschlager, A.; Kiess, W.; Bernhard, M.K.; Bertsche, T.; et al. Seizure disorders and developmental disorders: Impact on life of affected families—A structured interview. Eur. J. Pediatr. 2017, 176, 1121–1129. [Google Scholar] [CrossRef]
- Polder, J.J.; Meerding, W.J.; Bonneux, L.; van der Maas, P.J. Healthcare costs of intellectual disability in the Netherlands: A cost-of-illness perspective. J. Intellect. Disabil. Res. 2002, 46, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zigler, E. Mental retardation. Science 1967, 157, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Santen, G.W.; Clayton-Smith, J. The ARID1B phenotype: What we have learned so far. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Sadleir, L.G.; Mountier, E.I.; Gill, D.; Davis, S.; Joshi, C.; DeVile, C.; Kurian, M.A.; Mandelstam, S.; Wirrell, E.; Nickels, K.C.; et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: Early profound Thr226Met phenotype. Neurology 2017, 89, 1035–1042. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, L.; Ayub, M.; Vincent, J.B. The genetic basis of non-syndromic intellectual disability: A review. J. Neurodev. Disord. 2010, 2, 182–209. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Rudd, S.; Gratten, J.; Visscher, P.M.; Prins, J.B.; Dawson, P.A. Gene networks associated with non-syndromic intellectual disability. J. Neurogenet. 2018, 32, 6–14. [Google Scholar] [CrossRef]
- Ropers, H.H. Genetics of early onset cognitive impairment. Annu. Rev. Genom. Hum. Genet. 2010, 11, 161–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuchman, R. What is the Relationship between Autism Spectrum Disorders and Epilepsy? Semin. Pediatr. Neurol. 2017, 24, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Allison, C.; Wei, L.; Matthews, F.E.; Auyeung, B.; Wu, Y.Y.; Griffiths, S.; Zhang, J.; Baron-Cohen, S.; Brayne, C. Autism prevalence in China is comparable to Western prevalence. Mol. Autism 2019, 10, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jette, N. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology 2017, 88, 296–303. [Google Scholar] [CrossRef]
- Polanczyk, G.; de Lima, M.S.; Horta, B.L.; Biederman, J.; Rohde, L.A. The worldwide prevalence of ADHD: A systematic review and metaregression analysis. Am. J. Psychiatry 2007, 164, 942–948. [Google Scholar] [CrossRef]
- Thomas, R.; Sanders, S.; Doust, J.; Beller, E.; Glasziou, P. Prevalence of attention-deficit/hyperactivity disorder: A systematic review and meta-analysis. Pediatrics 2015, 135, e994–e1001. [Google Scholar] [CrossRef] [Green Version]
- Vissers, L.E.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef]
- Chiurazzi, P.; Pirozzi, F. Advances in understanding—Genetic basis of intellectual disability. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Lee, B.H.; Smith, T.; Paciorkowski, A.R. Autism spectrum disorder and epilepsy: Disorders with a shared biology. Epilepsy Behav. 2015, 47, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Mefford, H.C.; Batshaw, M.L.; Hoffman, E.P. Genomics, intellectual disability, and autism. N. Engl. J. Med. 2012, 366, 733–743. [Google Scholar] [CrossRef]
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Jacquemont, S.; Berry-Kravis, E.; Des Portes, V.; Stanfield, A.; Koumaras, B.; Rosenkranz, G.; Murgia, A.; Wolf, C.; Apostol, G.; et al. Mavoglurant in Fragile X Syndrome: Results of two open-label, extension trials in adults and adolescents. Sci. Rep. 2018, 8, 16970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Karnebeek, C.D.M. Evaluation of the Child With Developmental Impairments. Continuum 2018, 24, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Jamuar, S.S.; Tan, E.C. Clinical application of next-generation sequencing for Mendelian diseases. Hum. Genom. 2015, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevenon, J.; Duffourd, Y.; Masurel-Paulet, A.; Lefebvre, M.; Feillet, F.; El Chehadeh-Djebbar, S.; St-Onge, J.; Steinmetz, A.; Huet, F.; Chouchane, M.; et al. Diagnostic odyssey in severe neurodevelopmental disorders: Towards clinical whole-exome sequencing as a first-line diagnostic test. Clin. Genet. 2016, 89, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Stark, Z.; Schofield, D.; Martyn, M.; Rynehart, L.; Shrestha, R.; Alam, K.; Lunke, S.; Tan, T.Y.; Gaff, C.L.; White, S.M. Does genomic sequencing early in the diagnostic trajectory make a difference? A follow-up study of clinical outcomes and cost-effectiveness. Genet. Med. 2019, 21, 173–180. [Google Scholar] [CrossRef]
- Williams, J.C.; Barratt-Boyes, B.G.; Lowe, J.B. Supravalvular aortic stenosis. Circulation 1961, 24, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Ewart, A.K.; Morris, C.A.; Ensing, G.J.; Loker, J.; Moore, C.; Leppert, M.; Keating, M. A human vascular disorder, supravalvular aortic stenosis, maps to chromosome 7. Proc. Natl. Acad. Sci. USA 1993, 90, 3226–3230. [Google Scholar] [CrossRef] [Green Version]
- Ewart, A.K.; Morris, C.A.; Atkinson, D.; Jin, W.; Sternes, K.; Spallone, P.; Stock, A.D.; Leppert, M.; Keating, M.T. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat. Genet. 1993, 5, 11–16. [Google Scholar] [CrossRef]
- Noonan, J.A. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am. J. Dis. Child. 1968, 116, 373–380. [Google Scholar] [CrossRef]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C. Genotype to phenotype-discovery and characterization of novel genomic disorders in a “genotype-first” era. Genet. Med. 2009, 11, 836–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissers, L.; van Nimwegen, K.J.M.; Schieving, J.H.; Kamsteeg, E.J.; Kleefstra, T.; Yntema, H.G.; Pfundt, R.; van der Wilt, G.J.; Krabbenborg, L.; Brunner, H.G.; et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet. Med. 2017, 19, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Nimwegen, K.J.; Schieving, J.H.; Willemsen, M.A.; Veltman, J.A.; van der Burg, S.; van der Wilt, G.J.; Grutters, J.P. The diagnostic pathway in complex paediatric neurology: A cost analysis. Eur. J. Paediatr. Neurol. 2015, 19, 233–239. [Google Scholar] [CrossRef]
- Monroe, G.R.; Frederix, G.W.; Savelberg, S.M.; de Vries, T.I.; Duran, K.J.; van der Smagt, J.J.; Terhal, P.A.; van Hasselt, P.M.; Kroes, H.Y.; Verhoeven-Duif, N.M.; et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet. Med. 2016, 18, 949–956. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C. Chromosomes and Cytogenetics. 2014. Available online: https://www.nature.com/scitable/topic/chromosomes-and-cytogenetics-7 (accessed on 1 November 2022).
- Warkany, J.; Weinstein, E.D.; Soukup, S.W.; Rubinstein, J.H.; Curless, M.C. Chromosome Analyses in a Children’s Hospital. Selection of Patients and Results of Studies. Pediatrics 1964, 33, 290–305. [Google Scholar] [CrossRef]
- de Vries, B.B.; van den Ouweland, A.M.; Mohkamsing, S.; Duivenvoorden, H.J.; Mol, E.; Gelsema, K.; van Rijn, M.; Halley, D.J.; Sandkuijl, L.A.; Oostra, B.A.; et al. Screening and diagnosis for the fragile X syndrome among the mentally retarded: An epidemiological and psychological survey. Collaborative Fragile X Study Group. Am. J. Hum. Genet. 1997, 61, 660–667. [Google Scholar] [CrossRef] [Green Version]
- Ropers, H.H.; Hamel, B.C. X-linked mental retardation. Nat. Rev. Genet. 2005, 6, 46–57. [Google Scholar] [CrossRef]
- Neri, G.; Schwartz, C.E.; Lubs, H.A.; Stevenson, R.E. X-linked intellectual disability update 2017. Am. J. Med. Genet. A 2018, 176, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Breuning, M.H.; Dauwerse, H.G.; Fugazza, G.; Saris, J.J.; Spruit, L.; Wijnen, H.; Tommerup, N.; van der Hagen, C.B.; Imaizumi, K.; Kuroki, Y.; et al. Rubinstein-Taybi syndrome caused by submicroscopic deletions within 16p13.3. Am. J. Hum. Genet. 1993, 52, 249–254. [Google Scholar]
- Flint, J.; Wilkie, A.O.; Buckle, V.J.; Winter, R.M.; Holland, A.J.; McDermid, H.E. The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat. Genet. 1995, 9, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.J.; Regan, R.; Nicod, A.; Horsley, S.W.; Kearney, L.; Homfray, T.; Winter, R.M.; Bolton, P.; Flint, J. Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet 1999, 354, 1676–1681. [Google Scholar] [CrossRef] [PubMed]
- Ravnan, J.B.; Tepperberg, J.H.; Papenhausen, P.; Lamb, A.N.; Hedrick, J.; Eash, D.; Ledbetter, D.H.; Martin, C.L. Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J. Med. Genet. 2006, 43, 478–489. [Google Scholar] [CrossRef] [Green Version]
- De Vries, B.B.; Winter, R.; Schinzel, A.; van Ravenswaaij-Arts, C. Telomeres: A diagnosis at the end of the chromosomes. J. Med. Genet. 2003, 40, 385–398. [Google Scholar] [CrossRef] [Green Version]
- de Vries, B.B.; Pfundt, R.; Leisink, M.; Koolen, D.A.; Vissers, L.E.; Janssen, I.M.; Reijmersdal, S.; Nillesen, W.M.; Huys, E.H.; Leeuw, N.; et al. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005, 77, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, A.J.; Selzer, R.R.; Veltman, J.A.; Gimelli, S.; Gimelli, G.; Striano, P.; Coppola, A.; Regan, R.; Price, S.M.; Knoers, N.V.; et al. Characterization of a recurrent 15q24 microdeletion syndrome. Hum. Mol. Genet. 2007, 16, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Koolen, D.A.; Vissers, L.E.; Pfundt, R.; de Leeuw, N.; Knight, S.J.; Regan, R.; Kooy, R.F.; Reyniers, E.; Romano, C.; Fichera, M.; et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006, 38, 999–1001. [Google Scholar] [CrossRef]
- Xu, J. Next-Generation Sequencing, Current Technologies and Applications; Caister Academic Press: Poole, UK, 2014. [Google Scholar]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Redin, C.; Gerard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Tejada, M.I.; Gecz, J.; Shaw, M.; Corbett, M.; Haan, E.; Thompson, E.; Friend, K.; et al. Targeted Next-Generation Sequencing Analysis of 1,000 Individuals with Intellectual Disability. Hum. Mutat. 2015, 36, 1197–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- van der Sanden, B.; Schobers, G.; Galbany, J.C.; Koolen, D.A.; Sinnema, M.; van Reeuwijk, J.; Stumpel, C.; Kleefstra, T.; de Vries, B.B.A.; Ruiterkamp-Versteeg, M.; et al. The performance of genome sequencing as a first-tier test for neurodevelopmental disorders. Eur. J. Hum. Genet. 2022, 31, 81–88. [Google Scholar] [CrossRef]
- Sun, Y.; Ruivenkamp, C.A.; Hoffer, M.J.; Vrijenhoek, T.; Kriek, M.; van Asperen, C.J.; den Dunnen, J.T.; Santen, G.W. Next-generation diagnostics: Gene panel, exome, or whole genome? Hum. Mutat. 2015, 36, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Genome of the Netherlands, C. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014, 46, 818–825. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Vissers, L.E.; de Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; de Vries, P.; van Lier, B.; Arts, P.; Wieskamp, N.; del Rosario, M.; et al. A de novo paradigm for mental retardation. Nat. Genet. 2010, 42, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Wieczorek, D.; Graf, E.; Wieland, T.; Endele, S.; Schwarzmayr, T.; Albrecht, B.; Bartholdi, D.; Beygo, J.; Di Donato, N.; et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. Lancet 2012, 380, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; Han, Y.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Yuen, R.K.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef]
- Turner, T.N.; Hormozdiari, F.; Duyzend, M.H.; McClymont, S.A.; Hook, P.W.; Iossifov, I.; Raja, A.; Baker, C.; Hoekzema, K.; Stessman, H.A.; et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am. J. Hum. Genet. 2016, 98, 58–74. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.C.; Kim, G.E.; Pagnamenta, A.T.; Murakami, Y.; Carvill, G.L.; Meyer, E.; Copley, R.R.; Rimmer, A.; Barcia, G.; Fleming, M.R.; et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 2014, 23, 3200–3211. [Google Scholar] [CrossRef] [Green Version]
- Lubs, H.A. A marker X chromosome. Am. J. Hum. Genet. 1969, 21, 231–244. [Google Scholar] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Tzschach, A.; Grasshoff, U.; Beck-Woedl, S.; Dufke, C.; Bauer, C.; Kehrer, M.; Evers, C.; Moog, U.; Oehl-Jaschkowitz, B.; Di Donato, N.; et al. Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 2015, 23, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Schuurs-Hoeijmakers, J.H.; Vulto-van Silfhout, A.T.; Vissers, L.E.; Van De Vondervoort, I.I.; Van Bon, B.W.; De Ligt, J.; Gilissen, C.; Hehir-Kwa, J.Y.; Neveling, K.; Del Rosario, M.; et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J. Med. Genet. 2013, 50, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Santos-Cortez, R.L.P.; Khan, V.; Khan, F.S.; Mughal, Z.U.; Chakchouk, I.; Lee, K.; Rasheed, M.; Hamza, R.; Acharya, A.; Ullah, E.; et al. Novel candidate genes and variants underlying autosomal recessive neurodevelopmental disorders with intellectual disability. Hum. Genet. 2018, 137, 735–752. [Google Scholar] [CrossRef]
- Uher, R. The role of genetic variation in the causation of mental illness: An evolution-informed framework. Mol. Psychiatry 2009, 14, 1072–1082. [Google Scholar] [CrossRef] [Green Version]
- Veltman, I.M.; Veltman, J.A.; Arkesteijn, G.; Janssen, I.M.; Vissers, L.E.; de Jong, P.J.; van Kessel, A.G.; Schoenmakers, E.F. Chromosomal breakpoint mapping by arrayCGH using flow-sorted chromosomes. Biotechniques 2003, 35, 1066–1070. [Google Scholar] [CrossRef]
- Vulto-van Silfhout, A.T.; Hehir-Kwa, J.Y.; van Bon, B.W.; Schuurs-Hoeijmakers, J.H.; Meader, S.; Hellebrekers, C.J.; Thoonen, I.J.; de Brouwer, A.P.; Brunner, H.G.; Webber, C.; et al. Clinical significance of de novo and inherited copy-number variation. Hum. Mutat. 2013, 34, 1679–1687. [Google Scholar] [CrossRef]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef]
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Lelieveld, S.H.; Reijnders, M.R.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.J.; de Vries, P.; de Vries, B.B.; Willemsen, M.H.; Kleefstra, T.; Lohner, K.; et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Wilfert, A.B.; Sulovari, A.; Turner, T.N.; Coe, B.P.; Eichler, E.E. Recurrent de novo mutations in neurodevelopmental disorders: Properties and clinical implications. Genome Med. 2017, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Strachan, T.R.; Read, A. Human Molecular Genetics, 4th ed.; Taylor & Francis Inc.: Milton Park, UK, 2010. [Google Scholar]
- Porreca, G.J.; Zhang, K.; Li, J.B.; Xie, B.; Austin, D.; Vassallo, S.L.; LeProust, E.M.; Peck, B.J.; Emig, C.J.; Dahl, F.; et al. Multiplex amplification of large sets of human exons. Nat. Methods 2007, 4, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Turner, E.H.; Lee, C.; Ng, S.B.; Nickerson, D.A.; Shendure, J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat. Methods 2009, 6, 315–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Takata, A. Estimating contribution of rare non-coding variants to neuropsychiatric disorders. Psychiatry Clin. Neurosci. 2019, 73, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Turner, T.N.; Coe, B.P.; Dickel, D.E.; Hoekzema, K.; Nelson, B.J.; Zody, M.C.; Kronenberg, Z.N.; Hormozdiari, F.; Raja, A.; Pennacchio, L.A.; et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 2017, 171, 710–722.e712. [Google Scholar] [CrossRef] [Green Version]
- Krupp, D.R.; Barnard, R.A.; Duffourd, Y.; Evans, S.A.; Mulqueen, R.M.; Bernier, R.; Riviere, J.B.; Fombonne, E.; O’Roak, B.J. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 369–390. [Google Scholar] [CrossRef] [Green Version]
- Werling, D.M.; Brand, H.; An, J.Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef]
- Short, P.J.; McRae, J.F.; Gallone, G.; Sifrim, A.; Won, H.; Geschwind, D.H.; Wright, C.F.; Firth, H.V.; FitzPatrick, D.R.; Barrett, J.C.; et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 2018, 555, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.A.; Vissers, L.E.; Verbeek, M.M.; van Bon, B.W.; Geuer, S.; Gilissen, C.; Klepper, J.; Kwint, M.P.; Leen, W.G.; Pennings, M.; et al. Upstream SLC2A1 translation initiation causes GLUT1 deficiency syndrome. Eur. J. Hum. Genet. 2017, 25, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; Quaife, N.M.; Ramos-Hernandez, L.; Danecek, P.; Ferla, M.P.; Samocha, K.E.; Kaplanis, J.; Gardner, E.J.; Eberhardt, R.Y.; Chao, K.R.; et al. Non-coding region variants upstream of MEF2C cause severe developmental disorder through three distinct loss-of-function mechanisms. Am. J. Hum. Genet. 2021, 108, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Mefford, H.C. Poison exons in neurodevelopment and disease. Curr. Opin. Genet. Dev. 2020, 65, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Blakes, A.J.M.; Wai, H.A.; Davies, I.; Moledina, H.E.; Ruiz, A.; Thomas, T.; Bunyan, D.; Thomas, N.S.; Burren, C.P.; Greenhalgh, L.; et al. A systematic analysis of splicing variants identifies new diagnoses in the 100,000 Genomes Project. Genome Med. 2022, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Dawes, R.; Joshi, H.; Cooper, S.T. Empirical prediction of variant-activated cryptic splice donors using population-based RNA-Seq data. Nat. Commun. 2022, 13, 1655. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupianez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Melo, U.S.; Schopflin, R.; Acuna-Hidalgo, R.; Mensah, M.A.; Fischer-Zirnsak, B.; Holtgrewe, M.; Klever, M.K.; Turkmen, S.; Heinrich, V.; Pluym, I.D.; et al. Hi-C Identifies Complex Genomic Rearrangements and TAD-Shuffling in Developmental Diseases. Am. J. Hum. Genet. 2020, 106, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, K.; Yadav, R.; D’Haene, E.; Boone, P.M.; Erdin, S.; Gao, D.; Moyses-Oliveira, M.; Bhavsar, R.; Currall, B.B.; O’Keefe, K.; et al. Transcriptional and functional consequences of alterations to MEF2C and its topological organization in neuronal models. Am. J. Hum. Genet. 2022, 109, 2049–2067. [Google Scholar] [CrossRef]
- Redin, C.; Brand, H.; Collins, R.L.; Kammin, T.; Mitchell, E.; Hodge, J.C.; Hanscom, C.; Pillalamarri, V.; Seabra, C.M.; Abbott, M.A.; et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 2017, 49, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Merker, J.D.; Wenger, A.M.; Sneddon, T.; Grove, M.; Zappala, Z.; Fresard, L.; Waggott, D.; Utiramerur, S.; Hou, Y.; Smith, K.S.; et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 2018, 20, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauper, M.; Kucuk, E.; Wenger, A.M.; Chakraborty, S.; Baybayan, P.; Kwint, M.; van der Sanden, B.; Nelen, M.R.; Derks, R.; Brunner, H.G.; et al. Long-read trio sequencing of individuals with unsolved intellectual disability. Eur. J. Hum. Genet. 2021, 29, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Noyes, M.D.; Harvey, W.T.; Porubsky, D.; Sulovari, A.; Li, R.; Rose, N.R.; Audano, P.A.; Munson, K.M.; Lewis, A.P.; Hoekzema, K.; et al. Familial long-read sequencing increases yield of de novo mutations. Am. J. Hum. Genet. 2022, 109, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Levy-Sakin, M.; Pastor, S.; Mostovoy, Y.; Li, L.; Leung, A.K.Y.; McCaffrey, J.; Young, E.; Lam, E.T.; Hastie, A.R.; Wong, K.H.Y.; et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun. 2019, 10, 1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef]
- Sabatella, M.; Mantere, T.; Waanders, E.; Neveling, K.; Mensenkamp, A.R.; van Dijk, F.; Hehir-Kwa, J.Y.; Derks, R.; Kwint, M.; O’Gorman, L.; et al. Optical genome mapping identifies a germline retrotransposon insertion in SMARCB1 in two siblings with atypical teratoid rhabdoid tumors. J. Pathol. 2021, 255, 202–211. [Google Scholar] [CrossRef]
- de Bruijn, S.E.; Rodenburg, K.; Corominas, J.; Ben-Yosef, T.; Reurink, J.; Kremer, H.; Whelan, L.; Plomp, A.S.; Berger, W.; Farrar, G.J.; et al. Optical genome mapping and revisiting short-read genome sequencing data reveal previously overlooked structural variants disrupting retinal disease-associated genes. Genet. Med. 2022, 100345. [Google Scholar] [CrossRef]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O’Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017, 9, eaal5209. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Duffourd, Y.; Tisserant, E.; Relator, R.; Bruel, A.L.; Tran Mau-Them, F.; Denomme-Pichon, A.S.; Safraou, H.; Delanne, J.; Jean-Marcais, N.; et al. OMIXCARE: OMICS technologies solved about 33% of the patients with heterogeneous rare neuro-developmental disorders and negative exome sequencing results and identified 13% additional candidate variants. Front. Cell Dev. Biol. 2022, 10, 1021785. [Google Scholar] [CrossRef] [PubMed]
- Glinos, D.A.; Garborcauskas, G.; Hoffman, P.; Ehsan, N.; Jiang, L.; Gokden, A.; Dai, X.; Aguet, F.; Brown, K.L.; Garimella, K.; et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 2022, 608, 353–359. [Google Scholar] [CrossRef]
- Knudsen, T.B.; Spielmann, M.; Megason, S.G.; Faustman, E.M. Single-cell profiling for advancing birth defects research and prevention. Birth Defects Res. 2021, 113, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Larizza, L.; Finelli, P. Developmental disorders with intellectual disability driven by chromatin dysregulation: Clinical overlaps and molecular mechanisms. Clin. Genet. 2019, 95, 231–240. [Google Scholar] [CrossRef] [PubMed]
- van Bokhoven, H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011, 45, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Ciptasari, U.; van Bokhoven, H. The phenomenal epigenome in neurodevelopmental disorders. Hum. Mol. Genet. 2020, 29, R42–R50. [Google Scholar] [CrossRef] [PubMed]
- Choufani, S.; Cytrynbaum, C.; Chung, B.H.; Turinsky, A.L.; Grafodatskaya, D.; Chen, Y.A.; Cohen, A.S.; Dupuis, L.; Butcher, D.T.; Siu, M.T.; et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun. 2015, 6, 10207. [Google Scholar] [CrossRef] [Green Version]
- Kernohan, K.D.; Cigana Schenkel, L.; Huang, L.; Smith, A.; Pare, G.; Ainsworth, P.; Care4Rare Canada, C.; Boycott, K.M.; Warman-Chardon, J.; Sadikovic, B. Identification of a methylation profile for DNMT1-associated autosomal dominant cerebellar ataxia, deafness, narcolepsy. Clin. Epigenet. 2016, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, L.C.; Schwartz, C.; Skinner, C.; Rodenhiser, D.I.; Ainsworth, P.J.; Pare, G.; Sadikovic, B. Clinical Validation of Fragile X Syndrome Screening by DNA Methylation Array. J. Mol. Diagn. 2016, 18, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Sadikovic, B.; Aref-Eshghi, E.; Levy, M.A.; Rodenhiser, D. DNA methylation signatures in mendelian developmental disorders as a diagnostic bridge between genotype and phenotype. Epigenomics 2019, 11, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Rots, D.; Chater-Diehl, E.; Dingemans, A.J.M.; Goodman, S.J.; Siu, M.T.; Cytrynbaum, C.; Choufani, S.; Hoang, N.; Walker, S.; Awamleh, Z.; et al. Truncating SRCAP variants outside the Floating-Harbor syndrome locus cause a distinct neurodevelopmental disorder with a specific DNA methylation signature. Am. J. Hum. Genet. 2021, 108, 1053–1068. [Google Scholar] [CrossRef]
- Bend, E.G.; Aref-Eshghi, E.; Everman, D.B.; Rogers, R.C.; Cathey, S.S.; Prijoles, E.J.; Lyons, M.J.; Davis, H.; Clarkson, K.; Gripp, K.W.; et al. Gene domain-specific DNA methylation episignatures highlight distinct molecular entities of ADNP syndrome. Clin. Epigenet. 2019, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, P.; Hutton, T.J.; Allanson, J.E.; Buxton, B.; Campbell, L.E.; Clayton-Smith, J.; Donnai, D.; Karmiloff-Smith, A.; Metcalfe, K.; Murphy, K.C.; et al. Discriminating power of localized three-dimensional facial morphology. Am. J. Hum. Genet. 2005, 77, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferry, Q.; Steinberg, J.; Webber, C.; FitzPatrick, D.R.; Ponting, C.P.; Zisserman, A.; Nellaker, C. Diagnostically relevant facial gestalt information from ordinary photos. eLife 2014, 3, e02020. [Google Scholar] [CrossRef] [PubMed]
- van der Donk, R.; Jansen, S.; Schuurs-Hoeijmakers, J.H.M.; Koolen, D.A.; Goltstein, L.; Hoischen, A.; Brunner, H.G.; Kemmeren, P.; Nellaker, C.; Vissers, L.; et al. Next-generation phenotyping using computer vision algorithms in rare genomic neurodevelopmental disorders. Genet. Med. 2019, 21, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.M.; Kamphausen, S.B.; Zenker, M.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Bar-Haim, A.; Moosa, S.; Ehmke, N.; Gripp, K.W.; Pantel, J.T.; Danyel, M.; Mensah, M.A.; Horn, D.; Rosnev, S.; et al. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat. Genet. 2022, 54, 349–357. [Google Scholar] [CrossRef]
- Dingemans, A.J.M.; Hinne, M.; Truijen, K.M.G.; Goltstein, L.; van Reeuwijk, J.; de Leeuw, N.; Schuurs-Hoeijmakers, J.; Pfundt, R.; Diets, I.J.; den Hoed, J.; et al. PhenoScore: AI-based phenomics to quantify rare disease and genetic variation. medRxiv 2022. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jansen, S.; Vissers, L.E.L.M.; de Vries, B.B.A. The Genetics of Intellectual Disability. Brain Sci. 2023, 13, 231. https://doi.org/10.3390/brainsci13020231
Jansen S, Vissers LELM, de Vries BBA. The Genetics of Intellectual Disability. Brain Sciences. 2023; 13(2):231. https://doi.org/10.3390/brainsci13020231
Chicago/Turabian StyleJansen, Sandra, Lisenka E. L. M. Vissers, and Bert B. A. de Vries. 2023. "The Genetics of Intellectual Disability" Brain Sciences 13, no. 2: 231. https://doi.org/10.3390/brainsci13020231