
Pediatric Central Nervous System Tumor Overview and Emerging Treatment Considerations


Abstract
:1. Introduction
2. Materials and Methods
3. Pediatric-Type Diffuse Low-Grade Glioma (LGG)
3.1. Diffuse Astrocytoma, MYB- or MYBL1-Altered
3.2. Angiocentric Glioma
3.3. Polymorphous Low-Grade Neuroepithelial Tumor
3.4. Diffuse LGG, MAPK Pathway-Altered
3.5. Pilocytic Astrocytoma
3.6. Treatment
4. Pediatric-Type Diffuse High-Grade Gliomas (HGG)
4.1. DMG, H3 K27-Altered
4.2. Diffuse Pediatric HGG, H3-WT and IDH-WT
4.3. Diffuse Hemispheric Glioma (DHG), H3 G34-Mutant
4.4. Infant-Type Hemispheric Glioma
4.5. Treatment
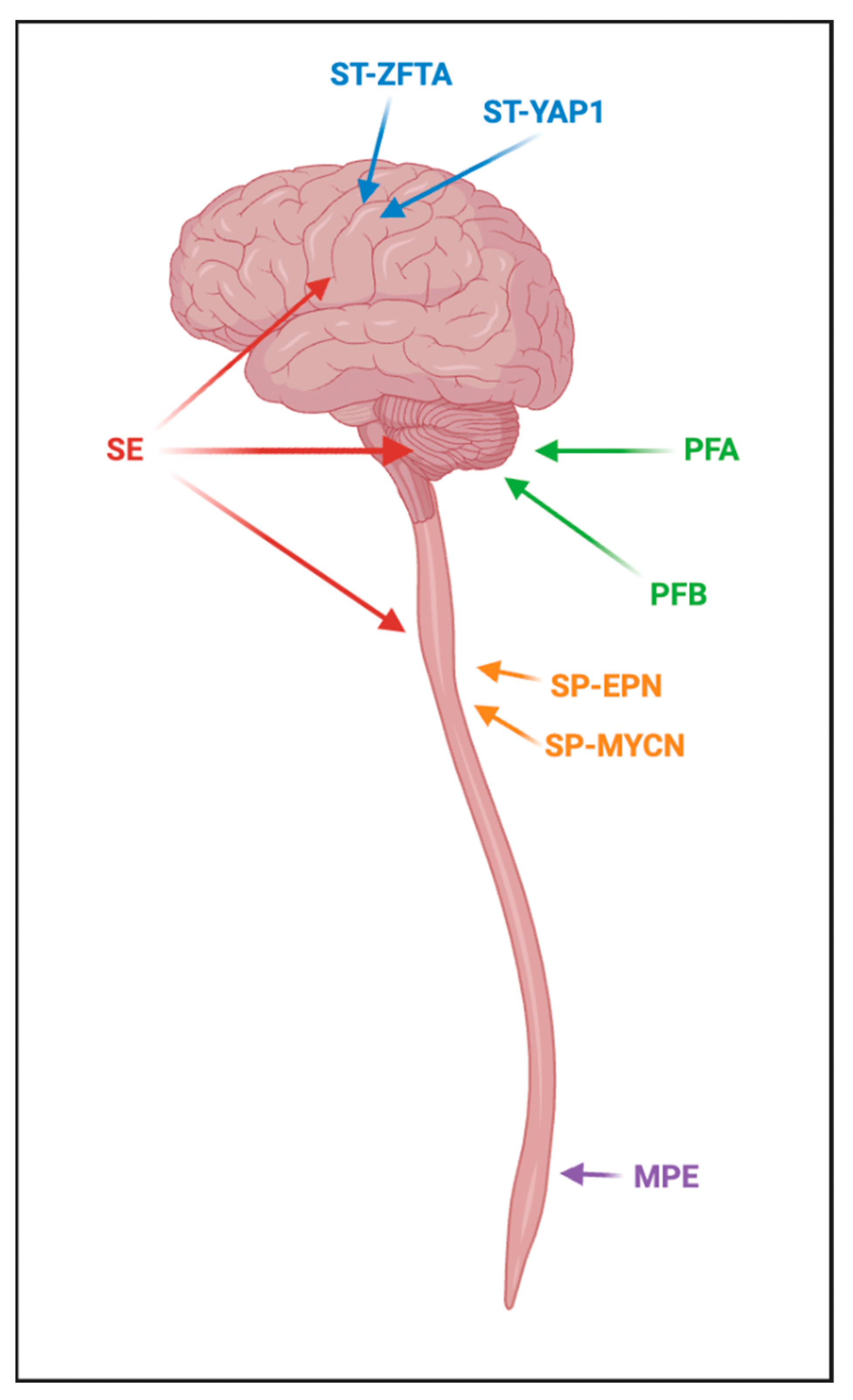
5. Ependymal Tumors
5.1. Supratentorial Ependymoma, ZFTA Fusion-Positive
5.2. Supratentorial Ependymoma, YAP1 Fusion-Positive
5.3. PFA Ependymoma
5.4. PFB Ependymoma
5.5. Treatment
6. CNS Embryonal Tumors
6.1. Medulloblastoma
6.1.1. Medulloblastoma, WNT-Activated
6.1.2. Medulloblastoma, SHH-Activated and TP53-WT
6.1.3. Medulloblastoma, SHH-Activated and TP53-Mutant
6.1.4. Medulloblastoma, Non-WNT/Non-SHH
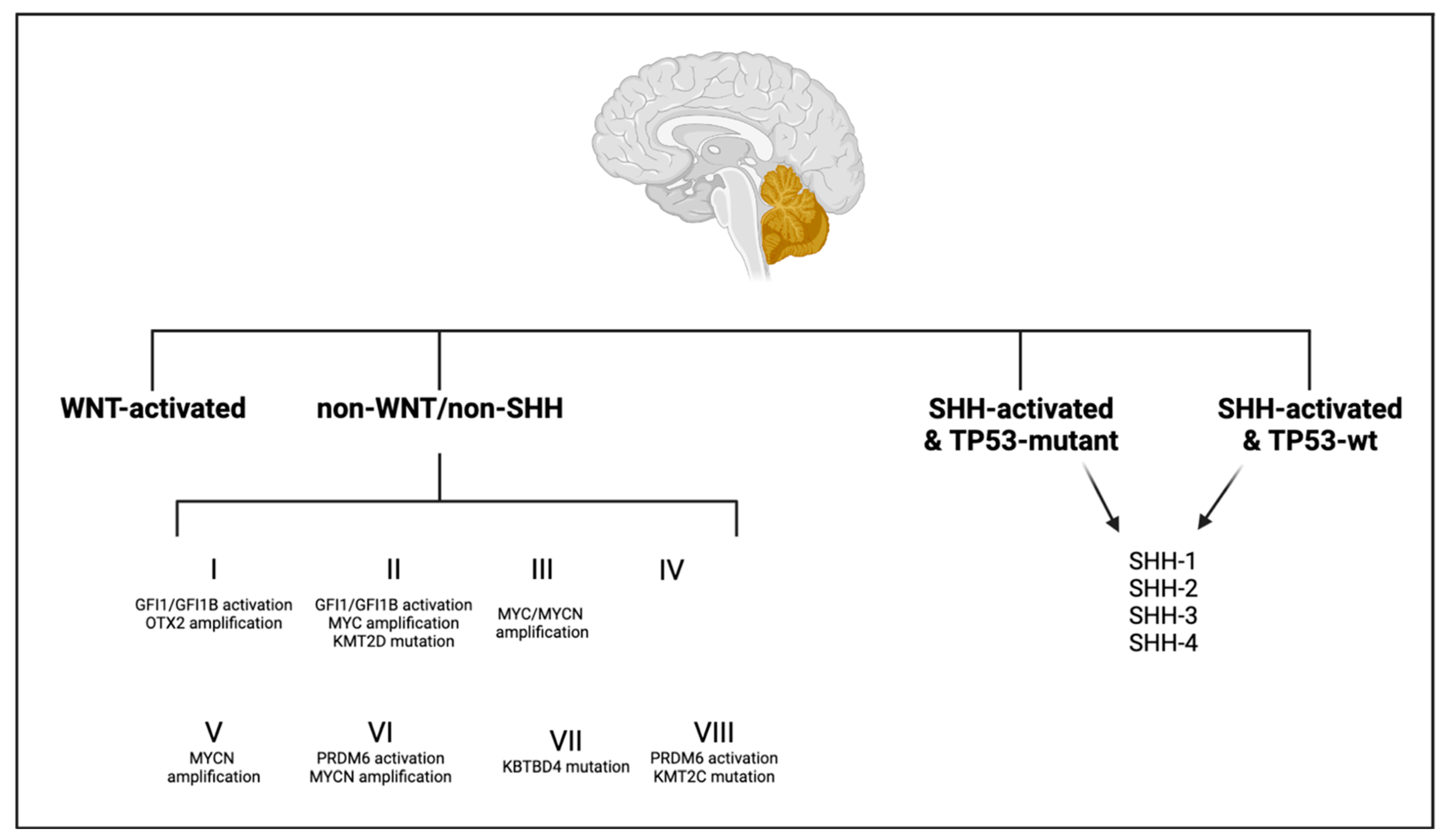
6.1.5. Treatment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, S.J.; Aw, N.M.Y.; Lim, M.J.R.; Tew Seow, W.; Low, D.C.Y.; Kimpo, M.S.; Ee Kar Tan, E.; Tsai Yeo, T.; Low, S.Y.Y.; Nga, V.D.W. Paediatric brain tumours in Singapore: A 15-year epidemiological and outcome study. J. Clin. Neurosci. 2022, 101, 154–161. [Google Scholar] [CrossRef]
- Subramanian, S.; Ahmad, T. Childhood Brain Tumors; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Pollack, I.F.; Agnihotri, S.; Broniscer, A. Childhood brain tumors: Current management, biological insights, and future directions. J. Neurosurg. Pediatr. 2019, 23, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.J.; Cullen, J.; Barnholtz-Sloan, J.S.; Ostrom, Q.T.; Langer, C.E.; Turner, M.C.; McKean-Cowdin, R.; Fisher, J.L.; Lupo, P.J.; Partap, S.; et al. Childhood brain tumor epidemiology: A brain tumor epidemiology consortium review. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2716–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adel Fahmideh, M.; Scheurer, M.E. Pediatric Brain Tumors: Descriptive Epidemiology, Risk Factors, and Future Directions. Cancer Epidemiol. Biomarkers Prev. 2021, 30, 813–821. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- de Blank, P.; Bandopadhayay, P.; Haas-Kogan, D.; Fouladi, M.; Fangusaro, J. Management of pediatric low-grade glioma. Curr. Opin. Pediatr. 2019, 31, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 2020, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Wefers, A.K.; Stichel, D.; Schrimpf, D.; Coras, R.; Pages, M.; Tauziede-Espariat, A.; Varlet, P.; Schwarz, D.; Soylemezoglu, F.; Pohl, U.; et al. Isomorphic diffuse glioma is a morphologically and molecularly distinct tumour entity with recurrent gene fusions of MYBL1 or MYB and a benign disease course. Acta Neuropathol. 2020, 139, 193–209. [Google Scholar] [CrossRef]
- Bale, T.A.; Rosenblum, M.K. The 2021 WHO Classification of Tumors of the Central Nervous System: An update on pediatric low-grade gliomas and glioneuronal tumors. Brain Pathol. 2022, 32, e13060. [Google Scholar] [CrossRef]
- Bandopadhayay, P.; Ramkissoon, L.A.; Jain, P.; Bergthold, G.; Wala, J.; Zeid, R.; Schumacher, S.E.; Urbanski, L.; O′Rourke, R.; Gibson, W.J.; et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat. Genet. 2016, 48, 273–282. [Google Scholar] [CrossRef]
- Buccoliero, A.M.; Caporalini, C.; Scagnet, M.; Mussa, F.; Giordano, F.; Sardi, I.; Migliastro, I.; Moscardi, S.; Conti, V.; Barba, C.; et al. Angiocentric glioma-associated seizures: The possible role of EATT2, pyruvate carboxylase and glutamine synthetase. Seizure 2021, 86, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Snuderl, M.; Jones, D.T.; Brathwaite, C.D.; Altman, N.; Lavi, E.; Saffery, R.; Sexton-Oates, A.; Blumcke, I.; Capper, D.; et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): An epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol. 2017, 133, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, X.; Zhao, J.; Wei, W.; Wang, W.; Kong, X.; Qian, R.; Niu, C.; Yao, Y. Clinical, Radiological, Pathological Features and Seizure Outcome With Surgical Management of Polymorphous Low-Grade Neuroepithelial Tumor of the Young Associated With Epilepsy. Front. Oncol. 2022, 12, 863373. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Kurokawa, R.; Capizzano, A.A.; Baba, A.; Ota, Y.; Pinarbasi, E.; Johnson, T.; Srinivasan, A.; Moritani, T. Neuroradiological features of the polymorphous low-grade neuroepithelial tumor of the young: Five new cases with a systematic review of the literature. Neuroradiology 2022, 64, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Khatua, S.; Wang, J.; Rajaram, V. Review of low-grade gliomas in children--evolving molecular era and therapeutic insights. Childs Nerv. Syst. 2015, 31, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Huse, J.T. The evolving molecular genetics of low-grade glioma. Adv. Anat. Pathol. 2015, 22, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, F.J.; Ligon, A.H.; Horkayne-Szakaly, I.; Rushing, E.J.; Ligon, K.L.; Vena, N.; Garcia, D.I.; Cameron, J.D.; Eberhart, C.G. BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J. Neuropathol. Exp. Neurol. 2012, 71, 789–794. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Salles, D.; Laviola, G.; Malinverni, A.C.M.; Stavale, J.N. Pilocytic Astrocytoma: A Review of General, Clinical, and Molecular Characteristics. J. Child. Neurol. 2020, 35, 852–858. [Google Scholar] [CrossRef]
- Hollon, T.; Hervey-Jumper, S.L.; Sagher, O.; Orringer, D.A. Advances in the Surgical Management of Low-Grade Glioma. Semin. Radiat. Oncol. 2015, 25, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.C.; Mehta, M.P. Low-Grade Glioma Radiotherapy Treatment and Trials. Neurosurg. Clin. N. Am. 2019, 30, 111–118. [Google Scholar] [CrossRef]
- Merchant, T.E.; Kun, L.E.; Wu, S.; Xiong, X.; Sanford, R.A.; Boop, F.A. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J. Clin. Oncol. 2009, 27, 3598–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ater, J.L.; Zhou, T.; Holmes, E.; Mazewski, C.M.; Booth, T.N.; Freyer, D.R.; Lazarus, K.H.; Packer, R.J.; Prados, M.; Sposto, R.; et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: A report from the Children′s Oncology Group. J. Clin. Oncol. 2012, 30, 2641–2647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassaletta, A.; Scheinemann, K.; Zelcer, S.M.; Hukin, J.; Wilson, B.A.; Jabado, N.; Carret, A.S.; Lafay-Cousin, L.; Larouche, V.; Hawkins, C.E.; et al. Phase II Weekly Vinblastine for Chemotherapy-Naive Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. J. Clin. Oncol. 2016, 34, 3537–3543. [Google Scholar] [CrossRef]
- Fangusaro, J.; Onar-Thomas, A.; Poussaint, T.Y.; Wu, S.; Ligon, A.H.; Lindeman, N.; Campagne, O.; Banerjee, A.; Gururangan, S.; Kilburn, L.B.; et al. A phase II trial of selumetinib in children with recurrent optic pathway and hypothalamic low-grade glioma without NF1: A Pediatric Brain Tumor Consortium study. Neuro Oncol. 2021, 23, 1777–1788. [Google Scholar] [CrossRef]
- Nicolaides, T.; Nazemi, K.J.; Crawford, J.; Kilburn, L.; Minturn, J.; Gajjar, A.; Gauvain, K.; Leary, S.; Dhall, G.; Aboian, M.; et al. Phase I study of vemurafenib in children with recurrent or progressive BRAF(V600E) mutant brain tumors: Pacific Pediatric Neuro-Oncology Consortium study (PNOC-002). Oncotarget 2020, 11, 1942–1952. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.J.; Gupta, N.; Hawkins, C.; et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro Oncol. 2017, 19, 153–161. [Google Scholar] [CrossRef] [Green Version]
- El-Ayadi, M.; Ansari, M.; Sturm, D.; Gielen, G.H.; Warmuth-Metz, M.; Kramm, C.M.; von Bueren, A.O. High-grade glioma in very young children: A rare and particular patient population. Oncotarget 2017, 8, 64564–64578. [Google Scholar] [CrossRef] [Green Version]
- Argersinger, D.P.; Rivas, S.R.; Shah, A.H.; Jackson, S.; Heiss, J.D. New Developments in the Pathogenesis, Therapeutic Targeting, and Treatment of H3K27M-Mutant Diffuse Midline Glioma. Cancers 2021, 13, 5280. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z. Oncohistone Mutations in Diffuse Intrinsic Pontine Glioma. Trends Cancer 2019, 5, 799–808. [Google Scholar] [CrossRef]
- Castel, D.; Kergrohen, T.; Tauziede-Espariat, A.; Mackay, A.; Ghermaoui, S.; Lechapt, E.; Pfister, S.M.; Kramm, C.M.; Boddaert, N.; Blauwblomme, T.; et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. 2020, 139, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Hohm, A.; Karremann, M.; Gielen, G.H.; Pietsch, T.; Warmuth-Metz, M.; Vandergrift, L.A.; Bison, B.; Stock, A.; Hoffmann, M.; Pham, M.; et al. Magnetic Resonance Imaging Characteristics of Molecular Subgroups in Pediatric H3 K27M Mutant Diffuse Midline Glioma. Clin. Neuroradiol. 2022, 32, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Johung, T.B.; Monje, M. Diffuse Intrinsic Pontine Glioma: New Pathophysiological Insights and Emerging Therapeutic Targets. Curr. Neuropharmacol. 2017, 15, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, H.; Pinches, A.; Picton, S.V.; Phillips, R.S. Survival rates and prognostic predictors of high grade brain stem gliomas in childhood: A systematic review and meta-analysis. J. Neurooncol. 2017, 135, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloum, R.; McConechy, M.K.; Mikael, L.G.; Fuller, C.; Drissi, R.; DeWire, M.; Nikbakht, H.; De Jay, N.; Yang, X.; Boue, D.; et al. Characterizing temporal genomic heterogeneity in pediatric high-grade gliomas. Acta Neuropathol. Commun. 2017, 5, 78. [Google Scholar] [CrossRef]
- Korshunov, A.; Schrimpf, D.; Ryzhova, M.; Sturm, D.; Chavez, L.; Hovestadt, V.; Sharma, T.; Habel, A.; Burford, A.; Jones, C.; et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017, 134, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Picart, T.; Barritault, M.; Poncet, D.; Berner, L.P.; Izquierdo, C.; Tabouret, E.; Figarella-Branger, D.; Idbaih, A.; Bielle, F.; Bourg, V.; et al. Characteristics of diffuse hemispheric gliomas, H3 G34-mutant in adults. Neurooncol. Adv. 2021, 3, vdab061. [Google Scholar] [CrossRef]
- Chen, K.Y.; Bush, K.; Klein, R.H.; Cervantes, V.; Lewis, N.; Naqvi, A.; Carcaboso, A.M.; Lechpammer, M.; Knoepfler, P.S. Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling. Commun. Biol. 2020, 3, 363. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.G.; Mueller, S.; Reddy, A.; Taylor, J.W.; Oberheim Bush, N.A.; Clarke, J.L.; Chang, S.M.; Gupta, N.; Berger, M.S.; Perry, A.; et al. Diffuse hemispheric glioma, H3 G34-mutant: Genomic landscape of a new tumor entity and prospects for targeted therapy. Neuro Oncol. 2021, 23, 1974–1976. [Google Scholar] [CrossRef]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Sahay, A.; Epari, S. Paediatric type diffuse high grade gliomas in the WHO CNS5 classification: What the pathologist needs to know? Indian. J. Pathol. Microbiol. 2022, 65, S50–S58. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood brain barrier: A challenge for effectual therapy of brain tumors. Biomed. Res. Int. 2015, 2015, 320941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metselaar, D.S.; du Chatinier, A.; Stuiver, I.; Kaspers, G.J.L.; Hulleman, E. Radiosensitization in Pediatric High-Grade Glioma: Targets, Resistance and Developments. Front. Oncol. 2021, 11, 662209. [Google Scholar] [CrossRef]
- Gallitto, M.; Lazarev, S.; Wasserman, I.; Stafford, J.M.; Wolden, S.L.; Terezakis, S.A.; Bindra, R.S.; Bakst, R.L. Role of Radiation Therapy in the Management of Diffuse Intrinsic Pontine Glioma: A Systematic Review. Adv. Radiat. Oncol. 2019, 4, 520–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatwin, H.V.; Cruz Cruz, J.; Green, A.L. Pediatric high-grade glioma: Moving toward subtype-specific multimodal therapy. FEBS J. 2021, 288, 6127–6141. [Google Scholar] [CrossRef] [PubMed]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Pre-Clinical Study of Panobinostat in Xenograft and Genetically Engineered Murine Diffuse Intrinsic Pontine Glioma Models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef] [Green Version]
- Homan, M.J.; Franson, A.; Ravi, K.; Roberts, H.; Pai, M.P.; Liu, C.; He, M.; Matvekas, A.; Koschmann, C.; Marini, B.L. Panobinostat penetrates the blood-brain barrier and achieves effective brain concentrations in a murine model. Cancer Chemother. Pharmacol. 2021, 88, 555–562. [Google Scholar] [CrossRef]
- Duchatel, R.J.; Mannan, A.; Woldu, A.S.; Hawtrey, T.; Hindley, P.A.; Douglas, A.M.; Jackson, E.R.; Findlay, I.J.; Germon, Z.P.; Staudt, D.; et al. Preclinical and clinical evaluation of German-sourced ONC201 for the treatment of H3K27M-mutant diffuse intrinsic pontine glioma. Neurooncol. Adv. 2021, 3, vdab169. [Google Scholar] [CrossRef]
- Ralff, M.D.; Lulla, A.R.; Wagner, J.; El-Deiry, W.S. ONC201: A new treatment option being tested clinically for recurrent glioblastoma. Transl. Cancer Res. 2017, 6, S1239–S1243. [Google Scholar] [CrossRef]
- Liu, X.; Hogg, G.D.; DeNardo, D.G. Rethinking immune checkpoint blockade: ′Beyond the T cell′. J. Immunother. Cancer 2021, 9, e001460. [Google Scholar] [CrossRef]
- Srinivas, S.; Bajpai, J. Immunotherapy in Special and Rare Situations: A Brief Review. J. Immunother. Precis. Oncol. 2021, 4, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Junger, S.T.; Timmermann, B.; Pietsch, T. Pediatric ependymoma: An overview of a complex disease. Childs Nerv. Syst. 2021, 37, 2451–2463. [Google Scholar] [CrossRef] [PubMed]
- Tauziede-Espariat, A.; Siegfried, A.; Nicaise, Y.; Kergrohen, T.; Sievers, P.; Vasiljevic, A.; Roux, A.; Dezamis, E.; Benevello, C.; Machet, M.C.; et al. Supratentorial non-RELA, ZFTA-fused ependymomas: A comprehensive phenotype genotype correlation highlighting the number of zinc fingers in ZFTA-NCOA1/2 fusions. Acta Neuropathol. Commun. 2021, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, S.A.; Robinson, G.W.; Onar-Thomas, A.; Orr, B.A.; Billups, C.A.; Bowers, D.C.; Bendel, A.E.; Hassall, T.; Crawford, J.R.; Partap, S.; et al. Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol. 2019, 21, 1319–1330. [Google Scholar] [CrossRef]
- Andreiuolo, F.; Varlet, P.; Tauziede-Espariat, A.; Junger, S.T.; Dorner, E.; Dreschmann, V.; Kuchelmeister, K.; Waha, A.; Haberler, C.; Slavc, I.; et al. Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: An entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol. 2019, 29, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Panwalkar, P.; Tamrazi, B.; Dang, D.; Chung, C.; Sweha, S.; Natarajan, S.K.; Pun, M.; Bayliss, J.; Ogrodzinski, M.P.; Pratt, D.; et al. Targeting integrated epigenetic and metabolic pathways in lethal childhood PFA ependymomas. Sci. Transl. Med. 2021, 13, eabc0497. [Google Scholar] [CrossRef]
- Araki, A.; Chocholous, M.; Gojo, J.; Dorfer, C.; Czech, T.; Heinzl, H.; Dieckmann, K.; Ambros, I.M.; Ambros, P.F.; Slavc, I.; et al. Chromosome 1q gain and tenascin-C expression are candidate markers to define different risk groups in pediatric posterior fossa ependymoma. Acta Neuropathol. Commun. 2016, 4, 88. [Google Scholar] [CrossRef] [Green Version]
- Zapotocky, M.; Beera, K.; Adamski, J.; Laperierre, N.; Guger, S.; Janzen, L.; Lassaletta, A.; Figueiredo Nobre, L.; Bartels, U.; Tabori, U.; et al. Survival and functional outcomes of molecularly defined childhood posterior fossa ependymoma: Cure at a cost. Cancer 2019, 125, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Kresbach, C.; Neyazi, S.; Schuller, U. Updates in the classification of ependymal neoplasms: The 2021 WHO Classification and beyond. Brain Pathol. 2022, 32, e13068. [Google Scholar] [CrossRef] [PubMed]
- Duffner, P.K.; Krischer, J.P.; Sanford, R.A.; Horowitz, M.E.; Burger, P.C.; Cohen, M.E.; Friedman, H.S.; Kun, L.E. Prognostic factors in infants and very young children with intracranial ependymomas. Pediatr. Neurosurg. 1998, 28, 215–222. [Google Scholar] [CrossRef]
- Robertson, P.L.; Zeltzer, P.M.; Boyett, J.M.; Rorke, L.B.; Allen, J.C.; Geyer, J.R.; Stanley, P.; Li, H.; Albright, A.L.; McGuire-Cullen, P.; et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: A report of the Children′s Cancer Group. J. Neurosurg. 1998, 88, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Miceli, R.; Giangaspero, F.; Boschetti, L.; Modena, P.; Antonelli, M.; Ferroli, P.; Bertin, D.; Pecori, E.; Valentini, L.; et al. Final results of the second prospective AIEOP protocol for pediatric intracranial ependymoma. Neuro Oncol. 2016, 18, 1451–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, T.E.; Li, C.; Xiong, X.; Kun, L.E.; Boop, F.A.; Sanford, R.A. Conformal radiotherapy after surgery for paediatric ependymoma: A prospective study. Lancet Oncol. 2009, 10, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Merchant, T.E.; Bendel, A.E.; Sabin, N.D.; Burger, P.C.; Shaw, D.W.; Chang, E.; Wu, S.; Zhou, T.; Eisenstat, D.D.; Foreman, N.K.; et al. Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J. Clin. Oncol. 2019, 37, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Junger, S.T.; Andreiuolo, F.; Mynarek, M.; Wohlers, I.; Rahmann, S.; Klein-Hitpass, L.; Dorner, E.; Zur Muhlen, A.; Velez-Char, N.; von Hoff, K.; et al. CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: A retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol. 2020, 140, 405–407. [Google Scholar] [CrossRef]
- Massimino, M.; Barretta, F.; Modena, P.; Witt, H.; Minasi, S.; Pfister, S.M.; Pajtler, K.W.; Antonelli, M.; Gandola, L.; Luisa Garre, M.; et al. Second series by the Italian Association of Pediatric Hematology and Oncology of children and adolescents with intracranial ependymoma: An integrated molecular and clinical characterization with a long-term follow-up. Neuro Oncol. 2021, 23, 848–857. [Google Scholar] [CrossRef]
- Van Mater, D.; Gururangan, S.; Becher, O.; Campagne, O.; Leary, S.; Phillips, J.J.; Huang, J.; Lin, T.; Poussaint, T.Y.; Goldman, S.; et al. A phase I trial of the CDK 4/6 inhibitor palbociclib in pediatric patients with progressive brain tumors: A Pediatric Brain Tumor Consortium study (PBTC-042). Pediatr. Blood Cancer 2021, 68, e28879. [Google Scholar] [CrossRef]
- Kram, D.E.; Henderson, J.J.; Baig, M.; Chakraborty, D.; Gardner, M.A.; Biswas, S.; Khatua, S. Embryonal Tumors of the Central Nervous System in Children: The Era of Targeted Therapeutics. Bioengineering 2018, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Suk, Y.; Gwynne, W.D.; Burns, I.; Venugopal, C.; Singh, S.K. Childhood Medulloblastoma: An Overview. Methods Mol. Biol. 2022, 2423, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.P.; Deepa, S.F.; Moinca, I.; Suresh, P.; Naidu, K.V. Medulloblastoma: A common pediatric tumor: Prognostic factors and predictors of outcome. Asian J. Neurosurg. 2015, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sursal, T.; Ronecker, J.S.; Dicpinigaitis, A.J.; Mohan, A.L.; Tobias, M.E.; Gandhi, C.D.; Jhanwar-Uniyal, M. Molecular Stratification of Medulloblastoma: Clinical Outcomes and Therapeutic Interventions. Anticancer. Res. 2022, 42, 2225–2239. [Google Scholar] [CrossRef]
- Cambruzzi, E. Medulloblastoma, WNT-activated/SHH-activated: Clinical impact of molecular analysis and histogenetic evaluation. Childs Nerv. Syst. 2018, 34, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Manoranjan, B.; Venugopal, C.; Bakhshinyan, D.; Adile, A.A.; Richards, L.; Kameda-Smith, M.M.; Whitley, O.; Dvorkin-Gheva, A.; Subapanditha, M.; Savage, N.; et al. Wnt activation as a therapeutic strategy in medulloblastoma. Nat. Commun. 2020, 11, 4323. [Google Scholar] [CrossRef]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic prognostication within medulloblastoma subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.Y.; Rosenblum, J.S.; Ho, W.S.; Heiss, J.D. New Developments in the Pathogenesis, Therapeutic Targeting, and Treatment of Pediatric Medulloblastoma. Cancers 2022, 14, 2285. [Google Scholar] [CrossRef]
- Shrestha, S.; Morcavallo, A.; Gorrini, C.; Chesler, L. Biological Role of MYCN in Medulloblastoma: Novel Therapeutic Opportunities and Challenges Ahead. Front. Oncol. 2021, 11, 694320. [Google Scholar] [CrossRef]
- Fults, D.W.; Taylor, M.D.; Garzia, L. Leptomeningeal dissemination: A sinister pattern of medulloblastoma growth. J. Neurosurg. Pediatr. 2019, 1–9. [Google Scholar] [CrossRef]
- Menyhart, O.; Gyorffy, B. Principles of tumorigenesis and emerging molecular drivers of SHH-activated medulloblastomas. Ann. Clin. Transl. Neurol. 2019, 6, 990–1005. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Murad, N.; Xu, Z.; Zhang, P.; Okonechnikov, K.; Kool, M.; Rivero-Hinojosa, S.; Lazarski, C.; Zheng, P.; Liu, Y.; et al. MYC Drives Group 3 Medulloblastoma through Transformation of Sox2(+) Astrocyte Progenitor Cells. Cancer Res. 2019, 79, 1967–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Dubuc, A.M.; Pfister, S.; Taylor, M.D. Molecular subgroups of medulloblastoma. Expert. Rev. Neurother. 2012, 12, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Noel, G. Medulloblastoma: Optimizing care with a multidisciplinary approach. J. Multidiscip. Healthc. 2019, 12, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Dhall, G.; O′Neil, S.H.; Ji, L.; Haley, K.; Whitaker, A.M.; Nelson, M.D.; Gilles, F.; Gardner, S.L.; Allen, J.C.; Cornelius, A.S.; et al. Excellent outcome of young children with nodular desmoplastic medulloblastoma treated on "Head Start" III: A multi-institutional, prospective clinical trial. Neuro Oncol. 2020, 22, 1862–1872. [Google Scholar] [CrossRef]
- Kian, W.; Roisman, L.C.; Goldstein, I.M.; Abo-Quider, A.; Samueli, B.; Wallach, N.; Alguayn, F.; Shalata, W.; Levitas, D.; Belochitski, O.; et al. Vismodegib as First-Line Treatment of Mutated Sonic Hedgehog Pathway in Adult Medulloblastoma. JCO Precis. Oncol. 2020, 4, PO.19.00264. [Google Scholar] [CrossRef]
- Nitta, R.T.; Bolin, S.; Luo, E.; Solow-Cordero, D.E.; Samghabadi, P.; Purzner, T.; Aujla, P.S.; Nwagbo, G.; Cho, Y.J.; Li, G. Correction: Casein kinase 2 inhibition sensitizes medulloblastoma to temozolomide. Oncogene 2020, 39, 2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Mulcahy Levy, J.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Kabir, T.F.; Kunos, C.A.; Villano, J.L.; Chauhan, A. Immunotherapy for Medulloblastoma: Current Perspectives. Immunotargets Ther. 2020, 9, 57–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Author Correction: Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2021, 27, 1117–1120. [Google Scholar] [CrossRef]
- Chang, C.H.; Housepian, E.M.; Herbert, C., Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 1969, 93, 1351–1359. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Study Title | NCT Number | Targeted Therapeutic Intervention | Country |
|---|---|---|---|
| DAY101 vs. Standard of Care Chemotherapy in Pediatric Patients with Low-Grade Glioma Requiring First-Line Systemic Therapy (LOGGIC/FIREFLY-2) | NCT05566795 | Drug: DAY101 | USA, Canada, Czechia, Korea, Switzerland |
| A Study to Evaluate DAY101 in Pediatric and Young Adult Patients with Relapsed or Progressive Low-Grade Glioma and Advance Solid Tumors | NCT04775485 | Drug: DAY101 | USA |
| SJ901: Evaluation of Mirdametinib in Children, Adolescents, and Young Adults with Low-Grade Glioma | NCT04923126 | Drug: Mirdametinib | USA |
| A Study of the Drugs Selumetinib Versus Carboplatin/ Vincristine in Patients with Neurofibromatosis and Low-Grade Glioma | NCT03871257 | Drug: Selumetinib Sulfate | USA, Canada |
| A Study of the Drugs Selumetinib vs. Carboplatin and Vincristine in Patients with Low-Grade Glioma | NCT04166409 | Drug: Selumetinib Sulfate | USA, Canada |
| A Study to Compare Treatment with the Drug Selumetinib Alone Versus Selumetinib and Vinblastine in Patients with Recurrent or Progressive Low-Grade Glioma | NCT04576117 | Drug: Selumetinib Sulfate | USA |
| Trametinib and Everolimus for Treatment of Pediatric and Young Adult Patients with Recurrent Gliomas | NCT04485559 | Drug: Everolimus Drug: Trametinib | USA |
| A Trial of Dabrafenib, Trametinib and Hydroxychloroquine for Patients with Recurrent LGG or HGG With a BRAF Aberration | NCT04201457 | Drug: Dabrafenib Drug: Trametinib | USA |
| Pediatric Low-Grade Glioma–MEK inhibitor Trial vs Chemotherapy | NCT05180825 | Drug: Trametinib | France |
| BGB-290 and Temozolomide in Treating Isocitrate Dehydrogenase (IDH)1/2-Mutant Grade I-IV Gliomas | NCT03749187 | Drug: PARP Inhibitor BGB-290 | USA |
| Pediatric Long-Term Follow-up and Rollover Study | NCT03975829 | Drug: Dabrafenib Drug: Trametinib | USA |
| Study Title | NCT Number | Immunotherapy Intervention | Country |
|---|---|---|---|
| Nivolumab in Combination with Temozolomide and Radiotherapy in Childrenand Adolescents with Newly Diagnosed High-grade Glioma | NCT04267146 | Drug: Nivolumab | France |
| Pembrolizumab in Treating Younger Patients with Recurrent, Progressive, or Refractory High-Grade Gliomas, Diffuse Intrinsic Pontine Gliomas, Hypermutated Brain Tumors, Ependymoma or Medulloblastoma | NCT02359565 | Drug: Pembrolizumab | USA, Canada |
| A Pilot Study of SurVaxMin Children Progressive or Relapsed Medulloblastoma, High Grade Glioma, Ependymoma and Newly Diagnosed Diffuse Intrinsic Pontine Glioma | NCT04978727 | Drug: SurVaxM | USA |
| REGN2810 in Pediatric Patients with Relapsed, Refractory Solid, or Central Nervous System (CNS) Tumors and Safety and Efficacy of REGN2810 in Combination with Radiotherapy in Pediatric Patients with Newly Diagnosed or Recurrent Glioma | NCT03690869 | Drug: Cemiplimab | USA |
| Autologous Dendritic Cells, Metronomic Cyclophosphamide and Checkpoint Blockade in Children with Relapsed HGG | NCT03879512 | Drug: Cancer vaccine and checkpoint blockade | Germany |
| C7R-GD2.CAR T Cells for Patients with GD2-expressing Brain Tumors (GAIL-B) | NCT04099797 | Drug: GD2-CART cells | USA |
| Loc3CAR: Locoregional Delivery of B7-H3-CAR T Cells for Pediatric Patients with Primary CNS Tumors | NCT05835687 | Drug: B7-H3 CART cells | USA |
| rHSC-DIPGVax Plus Checkpoint Blockade for the Treatment of Newly Diagnosed DIPG and DMG | NCT04943848 | Drug: rHSC- DIPGVax Drug: Balstilimab Drug: Zalifrelimab | USA |
| Stage | Extent of Disease |
|---|---|
| M0 | No evidence of subarachnoid or hematogenous metastasis |
| M1 | Microscopic tumor cells found in the CSF |
| M2 | Gross nodular seeding demonstrated in the cerebellar/cerebral subarachnoid space or in the third or lateral ventricles |
| M3 | Gross nodular seeding in the spinal subarachnoid space |
| M4 | Metastasis outside the cerebrospinal axis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damodharan, S.; Puccetti, D. Pediatric Central Nervous System Tumor Overview and Emerging Treatment Considerations. Brain Sci. 2023, 13, 1106. https://doi.org/10.3390/brainsci13071106
Damodharan S, Puccetti D. Pediatric Central Nervous System Tumor Overview and Emerging Treatment Considerations. Brain Sciences. 2023; 13(7):1106. https://doi.org/10.3390/brainsci13071106
Chicago/Turabian StyleDamodharan, Sudarshawn, and Diane Puccetti. 2023. "Pediatric Central Nervous System Tumor Overview and Emerging Treatment Considerations" Brain Sciences 13, no. 7: 1106. https://doi.org/10.3390/brainsci13071106
APA StyleDamodharan, S., & Puccetti, D. (2023). Pediatric Central Nervous System Tumor Overview and Emerging Treatment Considerations. Brain Sciences, 13(7), 1106. https://doi.org/10.3390/brainsci13071106