
Exploring Parkinson’s Disease-Associated Depression: Role of Inflammation on the Noradrenergic and Serotonergic Pathways

 ,
,
Abstract
:1. Introduction
2. The Interplay of Noradrenergic and Serotonergic Systems in Parkinson’s Disease
3. Inflammation in Parkinson’s Disease
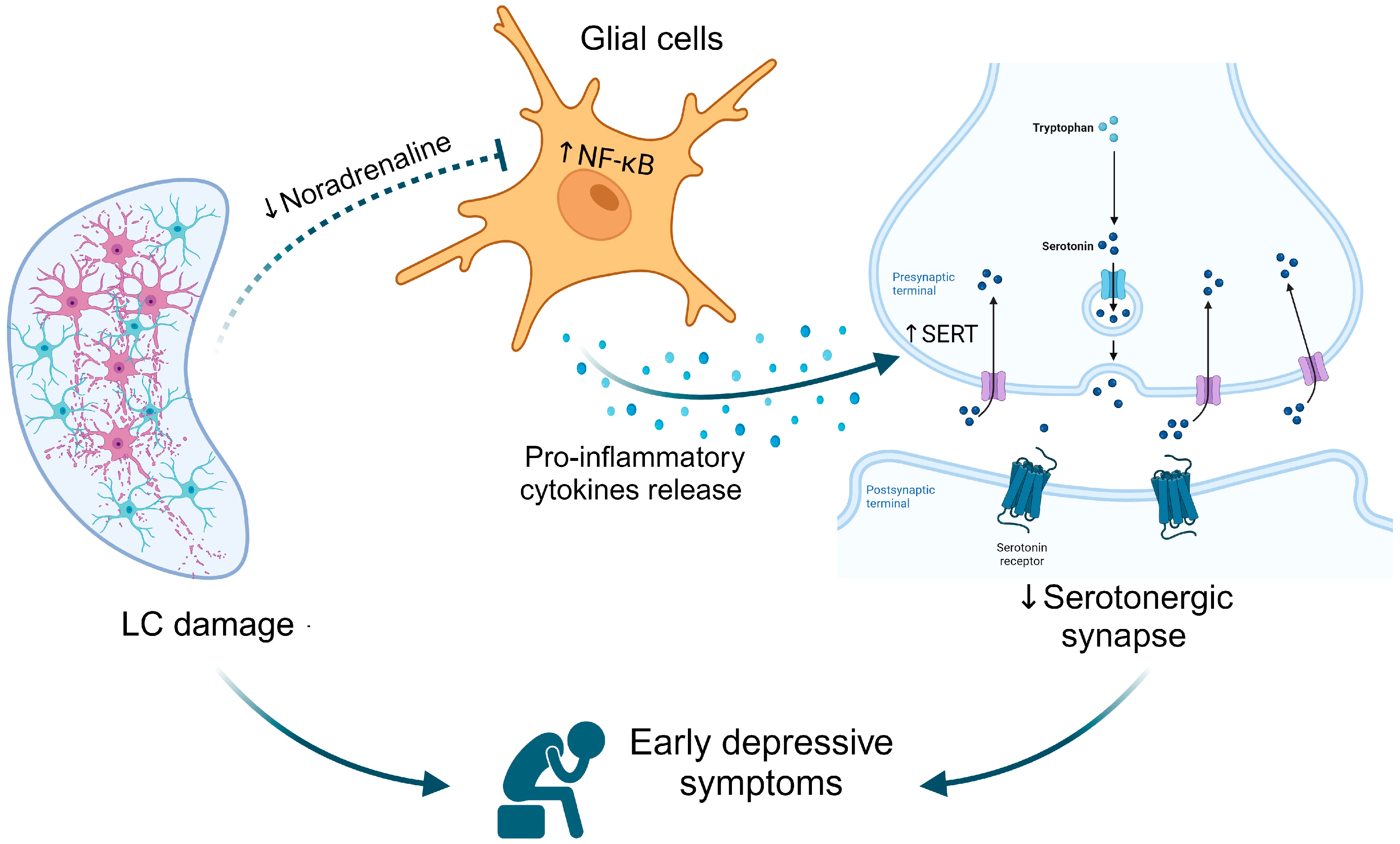
4. The Putative Interconnected Role of the Noradrenergic and Serotonergic Systems on Depression Associated with Parkinson’s Disease
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ahmad, M.H.; Rizvi, M.A.; Ali, M.; Mondal, A.C. Neurobiology of Depression in Parkinson’s Disease: Insights into Epidemiology, Molecular Mechanisms and Treatment Strategies. Ageing Res. Rev. 2023, 85, 101840. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Park. Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; de Souza, B.S.; Roversi, K.; Schuh, T.; Poli, A.; Takahashi, R.N.; Prediger, R.D. Temporal Development of Behavioral Impairments in Rats Following Locus Coeruleus Lesion Induced by 6-Hydroxydopamine: Involvement of Beta3-Adrenergic Receptors. Neuropharmacology 2019, 151, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Szot, P.; Franklin, A.; Miguelez, C.; Wang, Y.; Vidaurrazaga, I.; Ugedo, L.; Sikkema, C.; Wilkinson, C.W.; Raskind, M.A. Depressive-like behavior observed with a minimal loss of locus coeruleus (LC) neurons following administration of 6-hydroxydopamine is associated with electrophysiological changes and reversed with precursors of norepinephrine. Neuropharmacology 2016, 101, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, P.; Liu, C.; Xue, F.; Wang, Q.; Chen, Y.; Hou, R.; Chen, T. An investigation of neuromelanin distribution in substantia nigra and locus coeruleus in patients with Parkinson’s disease using neuromelanin-sensitive MRI. BMC Neurol. 2023, 23, 301. [Google Scholar] [CrossRef]
- Doppler, C.E.J.; Kinnerup, M.B.; Brune, C.; Farrher, E.; Betts, M.; Fedorova, T.D.; Schaldemose, J.L.; Knudsen, K.; Ismail, R.; Seger, A.D.; et al. Regional locus coeruleus degeneration is uncoupled from noradrenergic terminal loss in Parkinson’s disease. Brain 2021, 144, 2732–2744. [Google Scholar] [CrossRef]
- Bari, B.A.; Chokshi, V.; Schmidt, K. Locus coeruleus-norepinephrine: Basic functions and insights into Parkinson’s disease. Neural Regen. Res. 2020, 15, 1006–1013. [Google Scholar] [CrossRef]
- Giguere, N.; Nanni, S.B.; Trudeau, L.E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef]
- Dahl, M.J.; Kulesza, A.; Werkle-Bergner, M.; Mather, M. Declining locus coeruleus–dopaminergic and noradrenergic modulation of long-term memory in aging and Alzheimer’s disease. Neurosci. Biobehav. Rev. 2023, 153, 105358. [Google Scholar] [CrossRef] [PubMed]
- Fukada, K.; Endo, T.; Yokoe, M.; Hamasaki, T.; Hazama, T.; Sakoda, S. L-threo-3,4-dihydroxyphenylserine (L-DOPS) co-administered with entacapone improves freezing of gait in Parkinson’s disease. Med. Hypotheses 2013, 80, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, U. 6-hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 1968, 5, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; Marques, N.F.; Binder, L.B.; Tasca, C.I.; Prediger, R.D. Role of Prefrontal Cortex on Recognition Memory Deficits in Rats following 6-OHDA-Induced Locus coeruleus Lesion. Oxidative Med. Cell. Longev. 2020, 2020, 8324565. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The Mptp Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef]
- Shin, E.; Rogers, J.T.; Devoto, P.; Bjorklund, A.; Carta, M. Noradrenaline Neuron Degeneration Contributes to Motor Impairments and Development of L-Dopa-Induced Dyskinesia in a Rat Model of Parkinson’s Disease. Exp. Neurol. 2014, 257, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiao, Q.; Du, X.; Bi, M.; Han, S.; Jiao, L.; Jiang, H. Investigation of Behavioral Dysfunctions Induced by Monoamine Depletions in a Mouse Model of Parkinson’s Disease. Front. Cell Neurosci. 2018, 12, 241. [Google Scholar] [CrossRef]
- Thomas, B.; von Coelln, R.; Mandir, A.S.; Trinkaus, D.B.; Farah, M.H.; Lim, K.L.; Calingasan, N.Y.; Beal, M.F.; Dawson, V.L.; Dawson, T.M. MPTP and DSP-4 susceptibility of substantia nigra and locus coeruleus catecholaminergic neurons in mice is independent of parkin activity. Neurobiol. Dis. 2007, 26, 312–322. [Google Scholar] [CrossRef]
- Tritschler, L.; Gaillard, R.; Gardier, A.M.; David, D.J.; Guilloux, J.P. Consequences of the Monoaminergic Systems Cross-Talk in the Antidepressant Activity. Encephale 2018, 44, 264–273. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Kiferle, L.; Molloy, S.; Brooks, D.J.; Piccini, P. Staging of Serotonergic Dysfunction in Parkinson’s Disease: An in Vivo 11c-Dasb Pet Study. Neurobiol. Dis. 2010, 40, 216–221. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Gao, L.; Zhang, D.; Chen, L.; Wu, T. Common and Unique Dysconnectivity Profiles of Dorsal and Median Raphe in Parkinson’s Disease. Hum. Brain Mapp. 2023, 44, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Santiago, R.M.; Barbiero, J.; Gradowski, R.W.; Bochen, S.; Lima, M.M.; Da Cunha, C.; Andreatini, R.; Vital, M.A. Induction of depressive-like behavior by intranigral 6-OHDA is directly correlated with deficits in striatal dopamine and hippocampal serotonin. Behav. Brain Res. 2014, 259, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Kinoshita, K.-I.; Muroi, Y. Serotonin 5-HT4 Receptor Agonists Improve Facilitation of Contextual Fear Extinction in an MPTP-Induced Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5340. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, K.; Rahimmi, A. Oxidative Stress and Neuroinflammation in the Story of Parkinson’s Disease: Could Targeting These Pathways Write a Good Ending? J. Cell Physiol. 2018, 234, 23–32. [Google Scholar] [CrossRef]
- Furgiuele, A.; Pereira, F.C.; Martini, S.; Marino, F.; Cosentino, M. Dopaminergic Regulation of Inflammation and Immunity in Parkinson’s Disease: Friend or Foe? Clin. Transl. Immunol. 2023, 12, e1469. [Google Scholar] [CrossRef]
- Sampaio, T.B.; Sari, M.H.M.; Pesarico, A.P.; Nogueira, C.W. Delta-Aminolevulinate Dehydratase Activity Is Stimulated in a Mptp Mouse Model of Parkinson’s Disease: Correlation with Myeloperoxidase Activity. Cell Mol. Neurobiol. 2017, 37, 911–917. [Google Scholar] [CrossRef]
- Jurcau, A.; Andronie-Cioara, F.L.; Nistor-Cseppento, D.C.; Pascalau, N.; Rus, M.; Vasca, E.; Jurcau, M.C. The Involvement of Neuroinflammation in the Onset and Progression of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 14582. [Google Scholar] [CrossRef]
- Badanjak, K.; Fixemer, S.; Smajić, S.; Skupin, A.; Grünewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef]
- Doorn, K.J.; Goudriaan, A.; Blits-Huizinga, C.; Bol, J.G.; Rozemuller, A.J.; Hoogland, P.V.; Lucassen, P.J.; Drukarch, B.; van de Berg, W.D.; van Dam, A.M. Increased Amoeboid Microglial Density in the Olfactory Bulb of Parkinson’s and Alzheimer’s Patients. Brain Pathol. 2014, 24, 152–165. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In Vivo Imaging of Microglial Activation with [11c](R)-Pk11195 Pet in Idiopathic Parkinson’s Disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Lavisse, S.; Goutal, S.; Wimberley, C.; Tonietto, M.; Bottlaender, M.; Gervais, P.; Kuhnast, B.; Peyronneau, M.-A.; Barret, O.; Lagarde, J.; et al. Increased microglial activation in patients with Parkinson disease using [18F]-DPA714 TSPO PET imaging. Park. Relat. Disord. 2021, 82, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Stokholm, M.G.; Iranzo, A.; Østergaard, K.; Serradell, M.; Otto, M.; Svendsen, K.B.; Garrido, A.; Vilas, D.; Parbo, P.; Borghammer, P.; et al. Extrastriatal monoaminergic dysfunction and enhanced microglial activation in idiopathic rapid eye movement sleep behaviour disorder. Neurobiol. Dis. 2018, 115, 9–16. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Araujo, L.P.; Maricato, J.T.; Nascimento, V.M.; Guereschi, M.G.; Rezende, R.M.; Quintana, F.J.; Basso, A.S. Norepinephrine Controls Effector T Cell Differentiation through Beta2-Adrenergic Receptor-Mediated Inhibition of Nf-Kappab and Ap-1 in Dendritic Cells. J. Immunol. 2016, 196, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Yamaizumi, A.; Kawakami, A.; Islam, A.; Choudhury, M.E.; Takahashi, H.; Yano, H.; Tanaka, J. Anti-Inflammatory Effects of Noradrenaline on Lps-Treated Microglial Cells: Suppression of Nfkappab Nuclear Translocation and Subsequent Stat1 Phosphorylation. Neurochem. Int. 2015, 90, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Gavrilyuk, V.; Landreth, G.E.; O’Banion, M.K.; Weinberg, G.; Feinstein, D.L. Noradrenergic Depletion Increases Inflammatory Responses in Brain: Effects on Ikappab and Hsp70 Expression. J. Neurochem. 2003, 85, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Sastre, M.; Kreutz, A.; Gavrilyuk, V.; Klockgether, T.; Feinstein, D.L.; Heneka, M.T. Noradrenaline Induces Expression of Peroxisome Proliferator Activated Receptor Gamma (Ppargamma) in Murine Primary Astrocytes and Neurons. J. Neurochem. 2003, 86, 907–916. [Google Scholar] [CrossRef]
- Agac, D.; Estrada, L.D.; Maples, R.; Hooper, L.V.; Farrar, J.D. The Beta2-Adrenergic Receptor Controls Inflammation by Driving Rapid Il-10 Secretion. Brain Behav. Immun. 2018, 74, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Luong, K.; Nguyen, L.T. The Role of Beta-Adrenergic Blockers in Parkinson’s Disease: Possible Genetic and Cell-Signaling Mechanisms. Am. J. Alzheimer’s Dis. Other Demen 2013, 28, 306–317. [Google Scholar] [CrossRef]
- Mittal, S.; Bjornevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. Beta2-Adrenoreceptor Is a Regulator of the Alpha-Synuclein Gene Driving Risk of Parkinson’s Disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef]
- Malynn, S.; Campos-Torres, A.; Moynagh, P.; Haase, J. The Pro-Inflammatory Cytokine Tnf-Alpha Regulates the Activity and Expression of the Serotonin Transporter (Sert) in Astrocytes. Neurochem. Res. 2013, 38, 694–704. [Google Scholar] [CrossRef]
- Zhu, C.-B.; Lindler, K.M.; Owens, A.W.; Daws, L.C.; Blakely, R.D.; Hewlett, W.A. Interleukin-1 Receptor Activation by Systemic Lipopolysaccharide Induces Behavioral Despair Linked to MAPK Regulation of CNS Serotonin Transporters. Neuropsychopharmacology 2010, 35, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, R.K.; Bansal, Y.; Singh, R.; Saroj, P.; Bhandari, R.; Kumar, B.; Kuhad, A. Ido-1 Inhibition Protects against Neuroinflammation, Oxidative Stress and Mitochondrial Dysfunction in 6-Ohda Induced Murine Model of Parkinson’s Disease. Neurotoxicology 2021, 84, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; He, C.; Chen, J.; Li, J. Proton Magnetic Resonance Spectroscopy for the Early Diagnosis of Parkinson Disease in the Substantia Nigra and Globus Pallidus: A Meta-Analysis with Trial Sequential Analysis. Front. Neurol. 2022, 13, 838230. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Kalinderi, K.; Papaliagkas, V.; Fidani, L. Current Genetic Data on Depression and Anxiety in Parkinson’s Disease Patients. Park. Relat. Disord. 2023, 118, 105922. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Rajkumar, A.P.; Wan, Y.M.; Velayudhan, L.; Ffytche, D.; Chaudhuri, K.R.; Aarsland, D. Assessment and management of neuropsychiatric symptoms in Parkinson’s disease. CNS Drugs 2018, 32, 621–635. [Google Scholar] [CrossRef]
- Mayeux, R.; Stern, Y.; Cote, L.; Williams, J.B. Altered Serotonin Metabolism in Depressed Patients with Parkinson’s Disease. Neurology 1984, 34, 642–646. [Google Scholar] [CrossRef]
- Berg, D.; Supprian, T.; Hofmann, E.; Zeiler, B.; Jager, A.; Lange, K.W.; Reiners, K.; Becker, T.; Becker, G. Depression in Parkinson’s Disease: Brainstem Midline Alteration on Transcranial Sonography and Magnetic Resonance Imaging. J. Neurol. 1999, 246, 1186–1193. [Google Scholar] [CrossRef]
- Remy, P.; Doder, M.; Lees, A.; Turjanski, N.; Brooks, D. Depression in Parkinson’s Disease: Loss of Dopamine and Noradrenaline Innervation in the Limbic System. Brain 2005, 128, 1314–1322. [Google Scholar] [CrossRef]
- Frisina, P.G.; Haroutunian, V.; Libow, L.S. The Neuropathological Basis for Depression in Parkinson’s Disease. Park. Relat. Disord. 2009, 15, 144–148. [Google Scholar] [CrossRef]
- Ballanger, B.; Klinger, H.; Eche, J.; Lerond, J.; Vallet, A.E.; Le Bars, D.; Tremblay, L.; Sgambato-Faure, V.; Broussolle, E.; Thobois, S. Role of Serotonergic 1a Receptor Dysfunction in Depression Associated with Parkinson’s Disease. Mov. Disord. 2012, 27, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Qamhawi, Z.; Towey, D.; Shah, B.; Pagano, G.; Seibyl, J.; Marek, K.; Borghammer, P.; Brooks, D.J.; Pavese, N. Clinical correlates of raphe serotonergic dysfunction in early Parkinson’s disease. Brain 2015, 138, 2964–2973. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Krack, P.; Lhommée, E.; Météreau, E.; Klinger, H.; Favre, E.; Le Bars, D.; Schmitt, E.; Bichon, A.; Pelissier, P.; et al. The prominent role of serotonergic degeneration in apathy, anxiety and depression in de novo Parkinson’s disease. Brain 2016, 139, 2486–2502. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Liu, X.; Zhou, X.; Wang, H.; Zhou, W.; Jiang, J.; Peng, W.; Mo, L.; Tan, C.; Chen, L. Parkinson’s Disease-Related Risk of Suicide and Effect of Deep Brain Stimulation: Meta-Analysis. Park. Dis. 2020, 2020, 8091963. [Google Scholar] [CrossRef] [PubMed]
- Kocabicak, E.; Jahanshahi, A.; Schonfeld, L.; Hescham, S.-A.; Temel, Y.; Tan, S. Deep brain stimulation of the rat subthalamic nucleus induced inhibition of median raphe serotonergic and dopaminergic neurotransmission. Turk. Neurosurg. 2015, 25, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Alosaimi, F.; Temel, Y.; Hescham, S.; Witzig, V.S.; Almasabi, F.; Tan, S.K.H.; Jahanshahi, A. High-frequency stimulation of the subthalamic nucleus induces a sustained inhibition of serotonergic system via loss of cell phenotype. Sci. Rep. 2022, 12, 14011. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Yamada, S. The Relationships Among Metal Homeostasis, Mitochondria, and Locus Coeruleus in Psychiatric and Neurodegenerative Disorders: Potential Pathogenetic Mechanism and Therapeutic Implications. Cell. Mol. Neurobiol. 2023, 43, 963–989. [Google Scholar] [CrossRef]
- Liu, J.; Dong, J.; Wang, L.; Su, Y.; Yan, P.; Sun, S. Comparative Efficacy and Acceptability of Antidepressants in Parkinson’s Disease: A Network Meta-Analysis. PLoS ONE 2013, 8, e76651. [Google Scholar] [CrossRef]
- Devos, D.; Dujardin, K.; Poirot, I.; Moreau, C.; Cottencin, O.; Thomas, P.; Destee, A.; Bordet, R.; Defebvre, L. Comparison of Desipramine and Citalopram Treatments for Depression in Parkinson’s Disease: A Double-Blind, Randomized, Placebo-Controlled Study. Mov. Disord. 2008, 23, 850–857. [Google Scholar] [CrossRef]
- Menza, M.; Dobkin, R.D.; Marin, H.; Mark, M.H.; Gara, M.; Buyske, S.; Bienfait, K.; Dicke, A. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009, 72, 886–892. [Google Scholar] [CrossRef]
- Richard, I.H.; McDermott, M.P.; Kurlan, R.; Lyness, J.M.; Como, P.G.; Pearson, N.; Factor, S.A.; Juncos, J.; Ramos, C.S.; Brodsky, M.; et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology 2012, 78, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Altena, E.; Nombela, C.; Housden, C.R.; Maxwell, H.; Rittman, T.; Huddleston, C.; Rae, C.L.; Regenthal, R.; Sahakian, B.J.; et al. Selective serotonin reuptake inhibition modulates response inhibition in Parkinson’s disease. Brain 2014, 137, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tabu, H.; Ozaki, A.; Hamano, T.; Takeshima, T.; Reborn Study Group. Antidepressants for Depression, Apathy, and Gait Instability in Parkinson’s Disease: A Multicenter Randomized Study. Intern. Med. 2019, 58, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Stiedl, O.; Pappa, E.; Konradsson-Geuken, A.; Ogren, S.O. The role of the serotonin receptor subtypes 5-HT1A and 5-HT7 and its interaction in emotional learning and memory. Front. Pharmacol. 2015, 6, 162. [Google Scholar] [CrossRef]
- Thobois, S.; Prange, S.; Sgambato-Faure, V.; Tremblay, L.; Broussolle, E. Imaging the Etiology of Apathy, Anxiety, and Depression in Parkinson’s Disease: Implication for Treatment. Curr. Neurol. Neurosci. Rep. 2017, 17, 76. [Google Scholar] [CrossRef]
- Santiago, R.; Tonin, F.; Barbiero, J.; Zaminelli, T.; Boschen, S.; Andreatini, R.; Da Cunha, C.; Lima, M.; Vital, M. The nonsteroidal antiinflammatory drug piroxicam reverses the onset of depressive-like behavior in 6-OHDA animal model of Parkinson’s disease. Neuroscience 2015, 300, 246–253. [Google Scholar] [CrossRef]
- Zaminelli, T.; Gradowski, R.W.; Bassani, T.B.; Barbiero, J.K.; Santiago, R.M.; Maria-Ferreira, D.; Baggio, C.H.; Vital, M.A.B.F. Antidepressant and Antioxidative Effect of Ibuprofen in the Rotenone Model of Parkinson’s Disease. Neurotox. Res. 2014, 26, 351–362. [Google Scholar] [CrossRef]
- Campolo, M.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef]
- Vecchia, D.D.; Kanazawa, L.K.S.; Wendler, E.; de Almeida Soares Hocayen, P.; Bruginski, E.; Campos, F.R.; Stern, C.A.J.; Vital, M.; Miyoshi, E.; Wohr, M.; et al. Effects of Ketamine on Vocal Impairment, Gait Changes, and Anhedonia Induced by Bilateral 6-Ohda Infusion into the Substantia Nigra Pars Compacta in Rats: Therapeutic Implications for Parkinson’s Disease. Behav. Brain Res. 2018, 342, 1–10. [Google Scholar] [CrossRef]
- Vecchia, D.D.; Kanazawa, L.K.S.; Wendler, E.; Hocayen, P.A.S.; Vital, M.; Takahashi, R.N.; Da Cunha, C.; Miyoshi, E.; Andreatini, R. Ketamine Reversed Short-Term Memory Impairment and Depressive-Like Behavior in Animal Model of Parkinson’s Disease. Brain Res. Bull. 2021, 168, 63–73. [Google Scholar] [CrossRef]
- Holmes, S.E. Ketamine for the Treatment of Depression in Parkinson’s Disease (Ket-Pd). ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04944017 (accessed on 4 January 2024).
- Greenland, J.C.; Cutting, E.; Kadyan, S.; Bond, S.; Chhabra, A.; Williams-Gray, C.H. Azathioprine immunosuppression and disease modification in Parkinson’s disease (AZA-PD): A randomised double-blind placebo-controlled phase II trial protocol. BMJ Open 2020, 10, e040527. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Monaco, M.R.L.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the Exosomes in Parkinson Disease (Expand). Study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [PubMed]
- Santos-Garcia, D.; Mir, P.; Cubo, E.; Vela, L.; Rodriguez-Oroz, M.C.; Marti, M.J.; Arbelo, J.M.; Infante, J.; Kulisevsky, J.; Martinez-Martin, P.; et al. Coppadis-2015 (Cohort of Patients with Parkinson’s Disease in Spain, 2015), a Global--Clinical Evaluations, Serum Biomarkers, Genetic Studies and Neuroimaging--Prospective, Multicenter, Non-Interventional, Long-Term Study on Parkinson’s Disease Progression. BMC Neurol. 2016, 16, 26. [Google Scholar]

{kind=link}
| Reference | Neurotransmitter Systems | Subjects (n) | Methodology | Main Findings |
|---|---|---|---|---|
| Mayeux et al., 1984 [47] | Dopaminergic, noradrenergic, and serotonergic | In total, 16 depressed, 25 non-depressed PD patients, and 15 healthy controls | Measurement of 5-hydroxyindoleacetic acid (5-HIAA), homovanillic acid (HVA), and 3-methoxy-4-hydroxyphenylglycol (MHPG) in the cerebrospinal fluid (CSF) | 5-HIAA concentration, the main serotonin metabolite, was reduced in the CSF of PD patients with major depression and dysthymic disorder, as compared with that in non-depressed PD patients or controls. PD patients with major depression showed the greatest reduction in 5-HIAA levels in the CSF. The concentrations of HVA and MHPG in the CSF were not linked to the presence of depression. |
| Berg et al., 1999 [48] | Serotonergic | In total, 20 depressed and 11 non-depressed PD patients | Magnetic resonance imaging (MRI) and transcranial sonography (TCS) | Signal intensity reduction and T2 relaxation time of pontomesencephalic midline structures, which comprise the raphe nuclei (RN), in depressed PD patients as compared to those in non-depressed PD patients. No correlation was found between MRI signals or TCS echogenicity and motor symptoms or depression severity. |
| Remy et al., 2005 [49] | Dopaminergic and noradrenergic | In total, 8 depressed and 12 non-depressed PD patients | Positron emission tomography (PET) imaging | Depressed PD patients exhibited lower binding in the NET and DAT compared to those patients without depression, especially in the locus coeruleus (LC), anterior cingulate cortex, thalamus, amygdala, and ventral striatum. Anxiety severity in PD patients was found to be inversely correlated with the tracer binding in most of these regions, whereas apathy showed a specific inverse correlation in the ventral striatum. |
| Frisina et al., 2009 [50] | Dopaminergic, noradrenergic, and serotonergic | In total, 11 depressed and 9 non-depressed PD patients | Post-mortem pathological and immunohistochemical analysis | Pathological features were higher in the LC and dorsal vagus nerve of depressed PD patients as compared to those in non-depressed ones. Gliosis was more prevalent among depressed PD patients. No significant differences were identified in terms of gliosis and neuronal loss in the SN and RN of depressed and non-depressed PD patients. |
| Ballanger et al., 2012 [51] | Serotonergic | In total, 4 depressed, 8 non-depressed PD patients, and 7 healthy controls | PET imaging | Depressed PD patients demonstrated a reduction in the 5-HT1A receptor density as compared to non-depressed PD patients in the left hippocampus, right insula, left superior temporal cortex, and orbitofrontal cortex. Relative to healthy controls, non-depressed PD patients showed a bilateral decrease in the 5-HT1A receptor density in the inferior frontal cortex, right ventral striatum, and insula. In turn, depressed PD patients showed a specific 5-HT1A receptor decrease in the left dorsal anterior cingulate, orbitofrontal and temporal cortices, and right hippocampus. |
| Qamhawi et al., 2015 [52] | Serotonergic | In total, 345 early-onset PD patients, 56 putative PD patients without evidence of dopamine deficit, and 185 healthy controls | Single photon emission computed tomography (SPECT) | PD patients showed reduced availability of SERT in the raphe nuclei as compared to healthy controls and putative PD individuals without evidence of dopaminergic deficit. PD patients with resting tremors demonstrated an even more reduced availability of the SERT in the RN and less severe striatal dopaminergic deficits as compared to PD patients with akinetic–rigid syndromes and without tremors at rest. SERT availability in the RN was associated with resting tremor severity but not non-motor symptoms, including depression. |
| Maillet et al., 2016 [53] | Dopaminergic and serotonergic | In total, 15 Apathetic PD patients and 15 non-apathetic PD patients | PET imaging | Apathetic PD patients had higher depression and anxiety scores compared to non-apathetic PD ones. Non-apathetic PD patients showed mainly dopaminergic denervation in several regions, whereas those with apathy exhibited widespread dopaminergic and serotonergic degeneration in the striatum and thalamus. Specific dopaminergic disruption was reported in the SN–ventral tegmental area complex and specific serotonergic changes were found in the insula, orbitofrontal, and subgenual anterior cingulate cortices. Apathy severity was mainly correlated to specific serotonergic lesions in the right anterior area of the caudate nucleus and the orbitofrontal cortex. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampaio, T.B.; Schamne, M.G.; Santos, J.R.; Ferro, M.M.; Miyoshi, E.; Prediger, R.D. Exploring Parkinson’s Disease-Associated Depression: Role of Inflammation on the Noradrenergic and Serotonergic Pathways. Brain Sci. 2024, 14, 100. https://doi.org/10.3390/brainsci14010100
Sampaio TB, Schamne MG, Santos JR, Ferro MM, Miyoshi E, Prediger RD. Exploring Parkinson’s Disease-Associated Depression: Role of Inflammation on the Noradrenergic and Serotonergic Pathways. Brain Sciences. 2024; 14(1):100. https://doi.org/10.3390/brainsci14010100
Chicago/Turabian StyleSampaio, Tuane Bazanella, Marissa Giovanna Schamne, Jean Rodrigo Santos, Marcelo Machado Ferro, Edmar Miyoshi, and Rui Daniel Prediger. 2024. "Exploring Parkinson’s Disease-Associated Depression: Role of Inflammation on the Noradrenergic and Serotonergic Pathways" Brain Sciences 14, no. 1: 100. https://doi.org/10.3390/brainsci14010100
APA StyleSampaio, T. B., Schamne, M. G., Santos, J. R., Ferro, M. M., Miyoshi, E., & Prediger, R. D. (2024). Exploring Parkinson’s Disease-Associated Depression: Role of Inflammation on the Noradrenergic and Serotonergic Pathways. Brain Sciences, 14(1), 100. https://doi.org/10.3390/brainsci14010100