
Mitochondrial Dysfunction as a Biomarker of Illness State in Bipolar Disorder: A Critical Review
 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
Abstract
1. Bipolar Disorder
2. Mitochondrial Function
2.1. Mitochondria: Structure and Functions
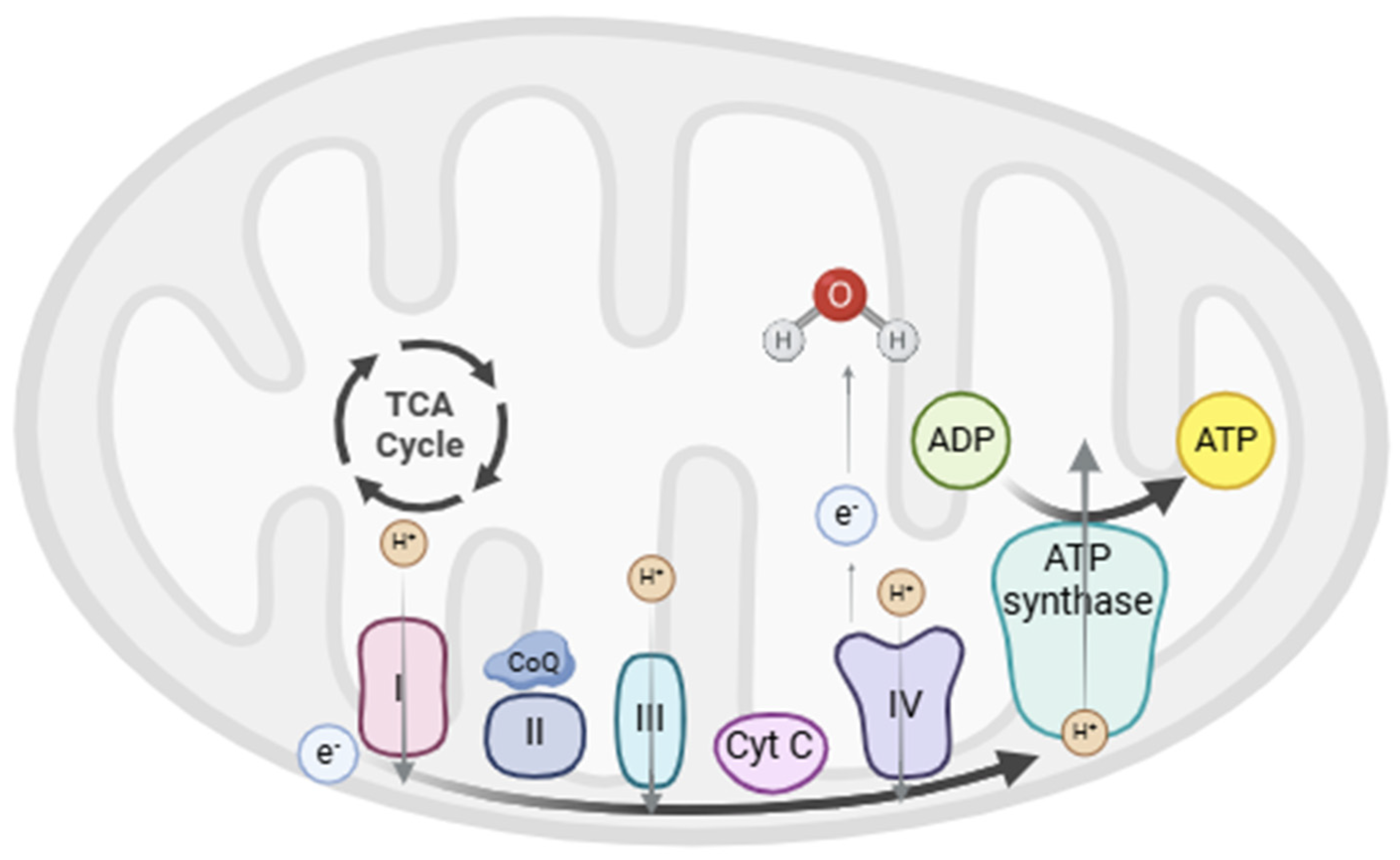
2.2. Oxidative Phosphorylation (OXPHOS)
2.3. Oxidative Stress
2.4. Other Functions of Mitochondria
3. Mitochondrial Dysfunction in Bipolar Disorder
3.1. Metabolic Changes
3.2. Electron Transport Chain (ETC)
3.2.1. Enzymatic Activity
3.2.2. Mitochondrial Respiratory Capacity
3.3. Oxidative Stress
3.4. Calcium Homeostasis
3.5. Mitochondrial Morphology and Dynamics
3.6. Mitochondrial Degradation and Apoptosis
3.7. Inflammation
3.8. Genetics
3.9. Mitochondrial DNA
3.10. Other Changes in Mitochondrial Function
4. Potential Mitochondrial-Related Therapies in Bipolar Disorder
4.1. Pharmacological Treatments
4.2. Nutrient Therapies
4.3. Physical Therapies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar Disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef]
- Vieta, E.; Berk, M.; Schulze, T.G.; Carvalho, A.F.; Suppes, T.; Calabrese, J.R.; Gao, K.; Miskowiak, K.W.; Grande, I. Bipolar Disorders. Nat. Rev. Dis. Primers 2018, 4, 18008. [Google Scholar] [CrossRef] [PubMed]
- Yatham, L.N.; Kennedy, S.H.; Parikh, S.V.; Schaffer, A.; Bond, D.J.; Frey, B.N.; Sharma, V.; Goldstein, B.I.; Rej, S.; Beaulieu, S.; et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) 2018 Guidelines for the Management of Patients with Bipolar Disorder. Bipolar Disord. 2018, 20, 97–170. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.F.; Firth, J.; Vieta, E. Bipolar Disorder. N. Engl. J. Med. 2020, 383, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Johansson, V.; Kuja-Halkola, R.; Cannon, T.D.; Hultman, C.M.; Hedman, A.M. A Population-Based Heritability Estimate of Bipolar Disorder—In a Swedish Twin Sample. Psychiatry Res. 2019, 278, 180–187. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, P.; Rijsdijk, F.; Andrew, M.; Sham, P.; Katz, R.; Cardno, A. The Heritability of Bipolar Affective Disorder and the Genetic Relationship to Unipolar Depression. Arch. Gen. Psychiatry 2003, 60, 497–502. [Google Scholar] [CrossRef]
- Aldinger, F.; Schulze, T.G. Environmental Factors, Life Events, and Trauma in the Course of Bipolar Disorder. Psychiatry Clin. Neurosci. 2017, 71, 6–17. [Google Scholar] [CrossRef]
- Menculini, G.; Balducci, P.M.; Attademo, L.; Bernardini, F.; Moretti, P.; Tortorella, A. Environmental Risk Factors for Bipolar Disorders and High-Risk States in Adolescence: A Systematic Review. Medicina 2020, 56, 689. [Google Scholar] [CrossRef]
- Chan, J.K.N.; Tong, C.C.H.Y.; Wong, C.S.M.; Chen, E.Y.H.; Chang, W.C. Life Expectancy and Years of Potential Life Lost in Bipolar Disorder: Systematic Review and Meta-Analysis. Br. J. Psychiatry 2022, 221, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Shao, L.; Sun, X.; Young, L.T. Increased Oxidative Stress in the Anterior Cingulate Cortex of Subjects with Bipolar Disorder and Schizophrenia. Bipolar Disord. 2009, 11, 523–529. [Google Scholar] [CrossRef]
- Morris, G.; Walder, K.; McGee, S.L.; Dean, O.M.; Tye, S.J.; Maes, M.; Berk, M. A Model of the Mitochondrial Basis of Bipolar Disorder. Neurosci. Biobehav. Rev. 2017, 74, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Romay-Tallon, R.; Brymer, K.J.; Caruncho, H.J.; Kalynchuk, L.E. Mitochondria and Mood: Mitochondrial Dysfunction as a Key Player in the Manifestation of Depression. Front. Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Kato, T. Neurobiological Basis of Bipolar Disorder: Mitochondrial Dysfunction Hypothesis and Beyond. Schizophr. Res. 2017, 187, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F. The Role of Mitochondrial Dysfunction in Psychiatric Disease. Dev. Disabil. Res. Rev. 2010, 16, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Fattal, O.; Budur, K.; Vaughan, A.J.; Franco, K. Review of the Literature on Major Mental Disorders in Adult Patients with Mitochondrial Diseases. Psychosomatics 2006, 47, 1–7. [Google Scholar] [CrossRef]
- Giménez-Palomo, A.; Dodd, S.; Anmella, G.; Carvalho, A.F.; Scaini, G.; Quevedo, J.; Pacchiarotti, I.; Vieta, E.; Berk, M. The Role of Mitochondria in Mood Disorders: From Physiology to Pathophysiology and to Treatment. Front. Psychiatry 2021, 12, 546801. [Google Scholar] [CrossRef]
- Pintus, F.; Floris, G.; Rufini, A. Nutrient Availability Links Mitochondria, Apoptosis, and Obesity. Aging 2012, 4, 734. [Google Scholar] [CrossRef]
- Caruso, G.; Benatti, C.; Blom, J.M.C.; Caraci, F.; Tascedda, F. The Many Faces of Mitochondrial Dysfunction in Depression: From Pathology to Treatment. Front. Pharmacol. 2019, 10, 995. [Google Scholar] [CrossRef]
- Budd, S.L.; Nicholls, D.G. Mitochondria in the Life and Death of Neurons. Essays Biochem. 1998, 33, 43–52. [Google Scholar] [CrossRef]
- Finkel, T. Radical Medicine: Treating Ageing to Cure Disease. Nat. Rev. Mol. Cell Biol. 2005, 6, 971–976. [Google Scholar] [CrossRef]
- Todorova, V.; Blokland, A. Mitochondria and Synaptic Plasticity in the Mature and Aging Nervous System. Curr. Neuropharmacol. 2016, 15, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.W.; Reynolds, I.J. Mitochondrial Trafficking and Morphology in Healthy and Injured Neurons. Prog. Neurobiol. 2006, 80, 241–268. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders—A Step towards Mitochondria Based Therapeutic Strategies. Biochim. Biophys. Acta 2017, 1863, 1066. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Kuhad, A. Mitochondrial Dysfunction in Depression. Curr. Neuropharmacol. 2016, 14, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Cuperfain, A.B.; Zhang, Z.L.; Kennedy, J.L.; Gonçalves, V.F. The Complex Interaction of Mitochondrial Genetics and Mitochondrial Pathways in Psychiatric Disease. Mol. Neuropsychiatry 2018, 4, 52–69. [Google Scholar] [CrossRef]
- Vélot, C.; Srere, P.A. Reversible Transdominant Inhibition of a Metabolic Pathway. In Vivo Evidence of Interaction between the Sequential Tricarboxylic Acid Cycle Enzymes in Yeast. J. Biol. Chem. 2000, 275, 12926–12933. [Google Scholar] [CrossRef]
- Davies, K.M.; Strauss, M.; Daum, B.; Kief, J.H.; Osiewacz, H.D.; Rycovska, A.; Zickermann, V.; Kühlbrandt, W. Macromolecular Organization of ATP Synthase and Complex I in Whole Mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 14121–14126. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef]
- Strauss, M.; Hofhaus, G.; Schröder, R.R.; Kühlbrandt, W. Dimer Ribbons of ATP Synthase Shape the Inner Mitochondrial Membrane. EMBO J. 2008, 27, 1154–1160. [Google Scholar] [CrossRef]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of Mitochondrial Oxidative Phosphorylation Supercomplexes and Mechanisms for Their Stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef]
- Wang, J.F. Defects of Mitochondrial Electron Transport Chain in Bipolar Disorder: Implications for Mood-Stabilizing Treatment. Can. J. Psychiatry 2007, 52, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. The Mitochondrial Production of Reactive Oxygen Species: Mechanisms and Implications in Human Pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Murphy, M.P. How Mitochondrial Damage Affects Cell Function. J. Biomed. Sci. 2002, 9, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free Radicals, Metals and Antioxidants in Oxidative Stress-Induced Cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative Diseases and Oxidative Stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, Oxidative Stress and the Biology of Ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial Quality Control: An Integrated Network of Pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef]
- Yoon, I.S.; Li, P.P.; Siu, K.P.; Kennedy, J.L.; Cooke, R.G.; Parikh, S.V.; Warsh, J.J. Altered IMPA2 Gene Expression and Calcium Homeostasis in Bipolar Disorder. Mol. Psychiatry 2001, 6, 678–683. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; del Arco, A.; Saheki, T.; Pardo, B.; Satrústegui, J. Calcium-Regulation of Mitochondrial Respiration Maintains ATP Homeostasis and Requires ARALAR/AGC1-Malate Aspartate Shuttle in Intact Cortical Neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef]
- Srivastava, R.; Faust, T.; Ramos, A.; Ishizuka, K.; Sawa, A. Dynamic Changes of the Mitochondria in Psychiatric Illnesses: New Mechanistic Insights From Human Neuronal Models. Biol. Psychiatry 2018, 83, 751–760. [Google Scholar] [CrossRef]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium Signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Duron, H.E.; Chaudhuri, D. Beyond the TCA Cycle: New Insights into Mitochondrial Calcium Regulation of Oxidative Phosphorylation. Biochem. Soc. Trans. 2023, 51, 1661. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Baron, K.T.; Wang, G.J.; Padua, R.A.; Campbell, C.; Thayer, S.A. NMDA-Evoked Consumption and Recovery of Mitochondrially Targeted Aequorin Suggests Increased Ca2+ Uptake by a Subset of Mitochondria in Hippocampal Neurons. Brain Res. 2003, 993, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Kato, T. Role of Mitochondrial DNA in Calcium Signaling Abnormality in Bipolar Disorder. Cell Calcium 2008, 44, 92–102. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The Pathophysiology of Mitochondrial Cell Death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Lindsten, T.; Zong, W.X.; Thompson, C.B. Defining the Role of the Bcl-2 Family of Proteins in the Nervous System. Neuroscientist 2005, 11, 10–15. [Google Scholar] [CrossRef]
- de Sousa, R.T.; Machado-Vieira, R.; Zarate, C.A.; Manji, H.K. Targeting Mitochondrially Mediated Plasticity to Develop Improved Therapeutics for Bipolar Disorder. Expert. Opin. Ther. Targets 2014, 18, 1131–1147. [Google Scholar] [CrossRef]
- Adzic, M.; Brkic, Z.; Bulajic, S.; Mitic, M.; Radojcic, M.B. Antidepressant Action on Mitochondrial Dysfunction in Psychiatric Disorders. Drug Dev. Res. 2016, 77, 400–406. [Google Scholar] [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and Age-Related Pathologies: Development of New Therapeutics by Targeting Mitochondrial Turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial Fusion, Fission, and Mitochondrial Toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Valadés, A.G.; Gonzalez-Franquesa, A.; Gama-Perez, P.; Claret, M.; Garcia-Roves, P.M. Emerging Concepts in Diabetes: Mitochondrial Dynamics and Glucose Homeostasis. Curr. Diabetes Rev. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- García-García, F.J.; Monistrol-Mula, A.; Cardellach, F.; Garrabou, G. Nutrition, Bioenergetics, and Metabolic Syndrome. Nutrients 2020, 12, 2785. [Google Scholar] [CrossRef] [PubMed]
- Burkhalter, J.; Fiumelli, H.; Allaman, I.; Chatton, J.-Y.; Martin, J.-L. Brain-Derived Neurotrophic Factor Stimulates Energy Metabolism in Developing Cortical Neurons. J. Neurosci. 2003, 23, 8212–8220. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Cameron, I.; Franklin, P.; Spedding, M. BDNF Increases Rat Brain Mitochondrial Respiratory Coupling at Complex I, but Not Complex II. Eur. J. Neurosci. 2004, 20, 1189–1196. [Google Scholar] [CrossRef]
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; Marin, L.; Charlton, M.P.; Atwood, H.L.; Zinsmaier, K.E. The GTPase DMiro Is Required for Axonal Transport of Mitochondria to Drosophila Synapses. Neuron 2005, 47, 379–393. [Google Scholar] [CrossRef]
- Verstreken, P.; Ly, C.V.; Venken, K.J.T.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic Mitochondria Are Critical for Mobilization of Reserve Pool Vesicles at Drosophila Neuromuscular Junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef]
- Toker, L.; Agam, G. Mitochondrial Dysfunction in Psychiatric Morbidity: Current Evidence and Therapeutic Prospects. Neuropsychiatr. Dis. Treat. 2015, 11, 2441–2447. [Google Scholar] [CrossRef]
- Scaini, G.; Rezin, G.T.; Carvalho, A.F.; Streck, E.L.; Berk, M.; Quevedo, J. Mitochondrial Dysfunction in Bipolar Disorder: Evidence, Pathophysiology and Translational Implications. Neurosci. Biobehav. Rev. 2016, 68, 694–713. [Google Scholar] [CrossRef]
- Kato, T.; Takahashi, S.; Shioiri, T.; Inubushi, T. Brain Phosphorous Metabolism in Depressive Disorders Detected by Phosphorus-31 Magnetic Resonance Spectroscopy. J. Affect. Disord. 1992, 26, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yuksel, C.; Chouinard, V.A.; Huynh, P.; Ryan, K.; Cohen, B.M.; Öngür, D. Abnormalities in High-Energy Phosphate Metabolism in First-Episode Bipolar Disorder Measured Using 31P-Magnetic Resonance Spectroscopy. Biol. Psychiatry 2018, 84, 797–802. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.L.; Naydenov, A.; Chu, M.; Matzilevich, D.; Konradi, C. Decrease in Creatine Kinase Messenger RNA Expression in the Hippocampus and Dorsolateral Prefrontal Cortex in Bipolar Disorder. Bipolar Disord. 2006, 8, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Dimick, M.K.; Sultan, A.; Duong, A.; Park, S.S.; El Soufi El Sabbagh, D.; Goldstein, B.I.; Andreazza, A.C. Peripheral Biomarkers of Mitochondrial Dysfunction in Adolescents with Bipolar Disorder. J. Psychiatr. Res. 2020, 123, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Zuccoli, G.S.; Saia-Cereda, V.M.; Nascimento, J.M.; Martins-de-Souza, D. The Energy Metabolism Dysfunction in Psychiatric Disorders Postmortem Brains: Focus on Proteomic Evidence. Front. Neurosci. 2017, 11, 493. [Google Scholar] [CrossRef]
- Dror, N.; Klein, E.; Karry, R.; Sheinkman, A.; Kirsh, Z.; Mazor, M.; Tzukerman, M.; Ben-Shachar, D. State-Dependent Alterations in Mitochondrial Complex I Activity in Platelets: A Potential Peripheral Marker for Schizophrenia. Mol. Psychiatry 2002, 7, 995–1001. [Google Scholar] [CrossRef]
- Taurines, R.; Thome, J.; Duvigneau, J.C.; Forbes-Robertson, S.; Yang, L.; Klampfl, K.; Romanos, J.; Müller, S.; Gerlach, M.; Mehler-Wex, C. Expression Analyses of the Mitochondrial Complex I 75-KDa Subunit in Early Onset Schizophrenia and Autism Spectrum Disorder: Increased Levels as a Potential Biomarker for Early Onset Schizophrenia. Eur. Child. Adolesc. Psychiatry 2010, 19, 441–448. [Google Scholar] [CrossRef]
- Peinado, J.R.; Diaz-Ruiz, A.; Frühbeck, G.; Malagon, M.M. Mitochondria in Metabolic Disease: Getting Clues from Proteomic Studies. Proteomics 2014, 14, 452–466. [Google Scholar] [CrossRef]
- Giménez-Palomo, A.; Guitart-Mampel, M.; Meseguer, A.; Borràs, R.; García-García, F.J.; Tobías, E.; Valls, L.; Alsina-Restoy, X.; Roqué, G.; Sánchez, E.; et al. Reduced Mitochondrial Respiratory Capacity in Patients with Acute Episodes of Bipolar Disorder: Could Bipolar Disorder Be a State-Dependent Mitochondrial Disease? Acta Psychiatr. Scand. 2024, 149, 52–64. [Google Scholar] [CrossRef]
- Giménez-Palomo, A.; Guitart-Mampel, M.; Roqué, G.; Sánchez, E.; Borràs, R.; Meseguer, A.; García-García, F.J.; Tobías, E.; Valls-Roca, L.; Anmella, G.; et al. Aerobic Capacity and Mitochondrial Function in Bipolar Disorder: A Longitudinal Study during Acute Phases and after Clinical Remission. Front. Psychiatry 2024, 15, 1386286. [Google Scholar] [CrossRef]
- Picard, M.; Prather, A.A.; Puterman, E.; Cuillerier, A.; Coccia, M.; Aschbacher, K.; Burelle, Y.; Epel, E.S. A Mitochondrial Health Index Sensitive to Mood and Caregiving Stress. Biol. Psychiatry 2018, 84, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Cordeiro, R.; Lima, C.C.; Fries, G.; Zunta-Soares, G.; Soares, J.C.; de Quevedo, J. Mitochondrial Health Index Correlates with Plasma Circulating Cell-Free Mitochondrial DNA in Bipolar Disorder. Res. Sq. 2023, 28, 4622–4631. [Google Scholar] [CrossRef]
- Andreazza, A.C.; Shao, L.; Wang, J.F.; Young, L.T. Mitochondrial Complex I Activity and Oxidative Damage to Mitochondrial Proteins in the Prefrontal Cortex of Patients with Bipolar Disorder. Arch. Gen. Psychiatry 2010, 67, 360–368, Erratum in Arch. Gen. Psychiatry 2010, 67, 1254. [Google Scholar] [CrossRef] [PubMed]
- Andreazza, A.C.; Kapczinski, F.; Kauer-Sant’Anna, M.; Walz, J.C.; Bond, D.J.; Gonçalves, C.A.; Young, L.T.; Yatham, L.N. 3-Nitrotyrosine and Glutathione Antioxidant System in Patients in the Early and Late Stages of Bipolar Disorder. J. Psychiatry Neurosci. 2009, 34, 263–271. [Google Scholar] [PubMed]
- Brown, N.C.; Andreazza, A.C.; Young, L.T. An Updated Meta-Analysis of Oxidative Stress Markers in Bipolar Disorder. Psychiatry Res. 2014, 218, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, J.F.; Tseng, M.; Young, L.T. Downregulation in Components of the Mitochondrial Electron Transport Chain in the Postmortem Frontal Cortex of Subjects with Bipolar Disorder. J. Psychiatry Neurosci. 2006, 31, 189–196. [Google Scholar]
- Benes, F.M.; Matzilevich, D.; Burke, R.E.; Walsh, J. The Expression of Proapoptosis Genes Is Increased in Bipolar Disorder, but Not in Schizophrenia. Mol. Psychiatry 2006, 11, 241–251. [Google Scholar] [CrossRef]
- Kuloglu, M.; Ustundag, B.; Atmaca, M.; Canatan, H.; Ertan Tezcan, A.; Cinkilinc, N. Lipid Peroxidation and Antioxidant Enzyme Levels in Patients with Schizophrenia and Bipolar Disorder. Cell Biochem. Funct. 2002, 20, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Ranjekar, P.K.; Hinge, A.; Hegde, M.V.; Ghate, M.; Kale, A.; Sitasawad, S.; Wagh, U.V.; Debsikdar, V.B.; Mahadik, S.P. Decreased Antioxidant Enzymes and Membrane Essential Polyunsaturated Fatty Acids in Schizophrenic and Bipolar Mood Disorder Patients. Psychiatry Res. 2003, 121, 109–122. [Google Scholar] [CrossRef]
- Savas, H.A.; Gergerlioglu, H.S.; Armutcu, F.; Herken, H.; Yilmaz, H.R.; Kocoglu, E.; Selek, S.; Tutkun, H.; Zoroglu, S.S.; Akyol, O. Elevated Serum Nitric Oxide and Superoxide Dismutase in Euthymic Bipolar Patients: Impact of Past Episodes. World J. Biol. Psychiatry 2006, 7, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Andreazza, A.C.; Viale, C.I.; Zanatto, V.; Cereser, V.; da Silva Vargas, R.; Kapczinski, F.; Portela, L.V.; Souza, D.O.; Salvador, M.; et al. Oxidative Stress Parameters in Unmedicated and Treated Bipolar Subjects during Initial Manic Episode: A Possible Role for Lithium Antioxidant Effects. Neurosci. Lett. 2007, 421, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Gergerlioglu, H.S.; Savas, H.A.; Bulbul, F.; Selek, S.; Uz, E.; Yumru, M. Changes in Nitric Oxide Level and Superoxide Dismutase Activity during Antimanic Treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Andreazza, A.C.; Cassini, C.; Rosa, A.R.; Leite, M.C.; de Almeida, L.M.V.; Nardin, P.; Cunha, A.B.N.; Ceresér, K.M.; Santin, A.; Gottfried, C.; et al. Serum S100B and Antioxidant Enzymes in Bipolar Patients. J. Psychiatr. Res. 2007, 41, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Trumbo, P.; Yates, A.A.; Schlicker, S.; Poos, M. Dietary Reference Intakes: Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. J. Am. Diet. Assoc. 2001, 101, 294–301. [Google Scholar] [CrossRef]
- de Sousa, R.T.; Zarate, C.A.; Zanetti, M.V.; Costa, A.C.; Talib, L.L.; Gattaz, W.F.; Machado-Vieira, R. Oxidative Stress in Early Stage Bipolar Disorder and the Association with Response to Lithium. J. Psychiatr. Res. 2014, 50, 36–41. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Uemura, T.; Green, M.; Corson, T.W.; Perova, T.; Li, P.P.; Warsh, J.J. Bcl-2 SNP Rs956572 Associates with Disrupted Intracellular Calcium Homeostasis in Bipolar I Disorder. Bipolar Disord. 2011, 13, 41–51. [Google Scholar] [CrossRef]
- O’Byrne, S.N.; Scott, J.W.; Pilotte, J.R.; da Santiago, A.S.; Langendorf, C.G.; Oakhill, J.S.; Eduful, B.J.; Couñago, R.M.; Wells, C.I.; Zuercher, W.J.; et al. In Depth Analysis of Kinase Cross Screening Data to Identify CaMKK2 Inhibitory Scaffolds. Molecules 2020, 25, 325. [Google Scholar] [CrossRef]
- Tokumitsu, H.; Iwabu, M.; Ishikawa, Y.; Kobayashi, R. Differential Regulatory Mechanism of Ca2+/Calmodulin-Dependent Protein Kinase Kinase Isoforms. Biochemistry 2001, 40, 13925–13932. [Google Scholar] [CrossRef] [PubMed]
- Atakhorrami, M.; Rahimi-Aliabadi, S.; Jamshidi, J.; Moslemi, E.; Movafagh, A.; Ohadi, M.; Mirabzadeh, A.; Emamalizadeh, B.; Ghaedi, H.; Gholipour, F.; et al. A Genetic Variant in CAMKK2 Gene Is Possibly Associated with Increased Risk of Bipolar Disorder. J. Neural Transm. 2016, 123, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Greenberg, M.E. From Synapse to Nucleus: Calcium-Dependent Gene Transcription in the Control of Synapse Development and Function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; McPhie, D.L.; Lange, N.T.; Punzell, S.; Elmiligy, S.; Ye, N.Z.; Froimowitz, M.P.; Hassinger, L.C.; Menesale, E.B.; Sargent, L.W.; et al. Abnormalities in Mitochondrial Structure in Cells from Patients with Bipolar Disorder. Am. J. Pathol. 2010, 177, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Safiulina, D.; Kaasik, A. Energetic and Dynamic: How Mitochondria Meet Neuronal Energy Demands. PLoS Biol. 2013, 11, e1001755. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Fries, G.R.; Valvassori, S.S.; Zeni, C.P.; Zunta-Soares, G.; Berk, M.; Soares, J.C.; Quevedo, J. Perturbations in the Apoptotic Pathway and Mitochondrial Network Dynamics in Peripheral Blood Mononuclear Cells from Bipolar Disorder Patients. Transl. Psychiatry 2017, 7, e1111. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Barichello, T.; Fries, G.R.; Kennon, E.A.; Andrews, T.; Nix, B.R.; Zunta-Soares, G.; Valvassori, S.S.; Soares, J.C.; Quevedo, J. TSPO Upregulation in Bipolar Disorder and Concomitant Downregulation of Mitophagic Proteins and NLRP3 Inflammasome Activation. Neuropsychopharmacology 2019, 44, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-W.; Rapoport, S.I.; Rao, J.S. Altered Expression of Apoptotic Factors and Synaptic Markers in Postmortem Brain from Bipolar Disorder Patients. Neurobiol. Dis. 2010, 37, 596–603. [Google Scholar] [CrossRef]
- Moutsatsou, P.; Tsoporis, J.N.; Salpeas, V.; Bei, E.; Alevizos, B.; Anagnostara, C.; Izhar, S.; Proteau, G.; Rizos, E.; Hatziagelaki, E.; et al. Peripheral Blood Lymphocytes from Patients with Bipolar Disorder Demonstrate Apoptosis and Differential Regulation of Advanced Glycation End Products and S100B. Clin. Chem. Lab. Med. 2014, 52, 999–1007. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt Phosphorylates HK-II at Thr-473 and Increases Mitochondrial HK-II Association to Protect Cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef]
- Liu, Y.; Ho, R.C.M.; Mak, A. Interleukin (IL)-6, Tumour Necrosis Factor Alpha (TNF-α) and Soluble Interleukin-2 Receptors (SIL-2R) Are Elevated in Patients with Major Depressive Disorder: A Meta-Analysis and Meta-Regression. J. Affect. Disord. 2012, 139, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Akundi, R.S. Mitochondria: A Connecting Link in the Major Depressive Disorder Jigsaw. Curr. Neuropharmacol. 2018, 17, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Norkett, R.; Modi, S.; Birsa, N.; Atkin, T.A.; Ivankovic, D.; Pathania, M.; Trossbach, S.V.; Korth, C.; Hirst, W.D.; Kittler, J.T. DISC1-Dependent Regulation of Mitochondrial Dynamics Controls the Morphogenesis of Complex Neuronal Dendrites. J. Biol. Chem. 2016, 291, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Michels, S.; Scheu, S.; Arolt, V.; Dannlowski, U.; Alferink, J. Mitochondria, Microglia, and the Immune System-How Are They Linked in Affective Disorders? Front. Psychiatry 2018, 9, 739. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, G.; Potash, J.B. New Developments in the Genetics of Bipolar Disorder. Curr. Psychiatry Rep. 2014, 16, 493. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Crane, D.E.; MacIntosh, B.J.; Young, L.T.; Arnold, P.; Ameis, S.; Goldstein, B.I. CACNA1C Rs1006737 Genotype and Bipolar Disorder: Focus on Intermediate Phenotypes and Cardiovascular Comorbidity. Neurosci. Biobehav. Rev. 2015, 55, 198–210. [Google Scholar] [CrossRef]
- Tachi, R.; Ohi, K.; Nishizawa, D.; Soda, M.; Fujikane, D.; Hasegawa, J.; Kuramitsu, A.; Takai, K.; Muto, Y.; Sugiyama, S.; et al. Mitochondrial Genetic Variants Associated with Bipolar Disorder and Schizophrenia in a Japanese Population. Int. J. Bipolar Disord. 2023, 11, 26. [Google Scholar] [CrossRef]
- Rollins, B.; Martin, M.V.; Sequeira, P.A.; Moon, E.A.; Morgan, L.Z.; Watson, S.J.; Schatzberg, A.; Akil, H.; Myers, R.M.; Jones, E.G.; et al. Mitochondrial Variants in Schizophrenia, Bipolar Disorder, and Major Depressive Disorder. PLoS ONE 2009, 4, e4913. [Google Scholar] [CrossRef] [PubMed]
- Munakata, K.; Tanaka, M.; Mori, K.; Washizuka, S.; Yoneda, M.; Tajima, O.; Akiyama, T.; Nanko, S.; Kunugi, H.; Tadokoro, K.; et al. Mitochondrial DNA 3644T-->C Mutation Associated with Bipolar Disorder. Genomics 2004, 84, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Mannu, P.; Saccaro, L.F.; Spera, V.; Cassano, P. Transcranial Photobiomodulation to Augment Lithium in Bipolar-I Disorder. Photobiomodulation Photomed. Laser Surg. 2019, 10, 577–578. [Google Scholar] [CrossRef]
- Kato, T.; Stine, O.C.; McMahon, F.J.; Crowe, R.R. Increased Levels of a Mitochondrial DNA Deletion in the Brain of Patients with Bipolar Disorder. Biol. Psychiatry 1997, 42, 871–875. [Google Scholar] [CrossRef]
- Shao, L.; Martin, M.V.; Watson, S.J.; Schatzberg, A.; Akil, H.; Myers, R.M.; Jones, E.G.; Bunney, W.E.; Vawter, M.P. Mitochondrial Involvement in Psychiatric Disorders. Ann. Med. 2008, 40, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Peltonen, L.; Paetau, A.; Leinonen, H.; Majander, A.; Somer, H. Inherited Idiopathic Dilated Cardiomyopathy with Multiple Deletions of Mitochondrial DNA. Lancet 1992, 340, 1319–1320. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Wigner, P.; Strycharz, J.; Swiderska, E.; Synowiec, E.; Szatkowska, M.; Sliwinska, A.; Talarowska, M.; Szemraj, J.; Su, K.-P.; et al. Mitochondrial DNA Copy Number, Damage, Repair and Degradation in Depressive Disorder. World J. Biol. Psychiatry 2019, 21, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, N.; Otsuka, I.; Numata, S.; Yanagi, M.; Mouri, K.; Okazaki, S.; Boku, S.; Horai, T.; Ohmori, T.; Shirakawa, O.; et al. Mitochondrial DNA Copy Number of Peripheral Blood in Bipolar Disorder: The Present Study and a Meta-Analysis. Psychiatry Res. 2018, 269, 115–117. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Tai, P.-A.; Poly, T.N.; Islam, M.M.; Yang, H.-C.; Wu, C.-C.; Li, Y.-C. Increased Risk of Dementia in Patients with Antidepressants: A Meta-Analysis of Observational Studies. Behav. Neurol. 2018, 2018, 5315098. [Google Scholar] [CrossRef]
- Ceylan, D.; Karacicek, B.; Tufekci, K.U.; Aksahin, I.C.; Senol, S.H.; Genc, S. Mitochondrial DNA Oxidation, Methylation, and Copy Number Alterations in Major and Bipolar Depression. Front. Psychiatry 2023, 14, 1304660. [Google Scholar] [CrossRef]
- Kageyama, Y.; Kasahara, T.; Kato, M.; Sakai, S.; Deguchi, Y.; Tani, M.; Kuroda, K.; Hattori, K.; Yoshida, S.; Goto, Y.; et al. The Relationship between Circulating Mitochondrial DNA and Inflammatory Cytokines in Patients with Major Depression. J. Affect. Disord. 2018, 233, 15–20. [Google Scholar] [CrossRef]
- Chang, C.-C.; Jou, S.-H.; Lin, T.-T.; Liu, C.-S. Mitochondrial DNA Variation and Increased Oxidative Damage in Euthymic Patients with Bipolar Disorder. Psychiatry Clin. Neurosci. 2014, 68, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Plein, H.; Ferreira, D.; Jersky, B. Blunted Adenosine A2a Receptor Function in Platelets in Patients with Major Depression. Eur. Neuropsychopharmacol. 2001, 11, 183–186. [Google Scholar] [CrossRef]
- Adzic, M.; Lukic, I.; Mitic, M.; Djordjevic, J.; Elaković, I.; Djordjevic, A.; Krstic-Demonacos, M.; Matić, G.; Radojcic, M. Brain Region- and Sex-Specific Modulation of Mitochondrial Glucocorticoid Receptor Phosphorylation in Fluoxetine Treated Stressed Rats: Effects on Energy Metabolism. Psychoneuroendocrinology 2013, 38, 2914–2924. [Google Scholar] [CrossRef]
- Madireddy, S.; Madireddy, S. Therapeutic Interventions to Mitigate Mitochondrial Dysfunction and Oxidative Stress-Induced Damage in Patients with Bipolar Disorder. Int. J. Mol. Sci. 2022, 23, 1844. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Andrews, T.; Lima, C.N.C.; Benevenuto, D.; Streck, E.L.; Quevedo, J. Mitochondrial Dysfunction as a Critical Event in the Pathophysiology of Bipolar Disorder. Mitochondrion 2021, 57, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Clay, H.B.; Sillivan, S.; Konradi, C. Mitochondrial Dysfunction and Pathology in Bipolar Disorder and Schizophrenia. Int. J. Dev. Neurosci. 2011, 29, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shao, L.; Young, L.T.; Wang, J.F. Role of Glutathione in Neuroprotective Effects of Mood Stabilizing Drugs Lithium and Valproate. Neuroscience 2007, 144, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Manji, H.K.; Zarate, C.A. The Role of Lithium in the Treatment of Bipolar Disorder: Convergent Evidence for Neurotrophic Effects as a Unifying Hypothesis. Bipolar Disord. 2009, 11 (Suppl. S2), 92–109. [Google Scholar] [CrossRef] [PubMed]
- Valvassori, S.S.; Rezin, G.T.; Ferreira, C.L.; Moretti, M.; Gonçalves, C.L.; Cardoso, M.R.; Streck, E.L.; Kapczinski, F.; Quevedo, J. Effects of Mood Stabilizers on Mitochondrial Respiratory Chain Activity in Brain of Rats Treated with D-Amphetamine. J. Psychiatr. Res. 2010, 44, 903–909. [Google Scholar] [CrossRef]
- Maurer, I.C.; Schippel, P.; Volz, H.-P. Lithium-Induced Enhancement of Mitochondrial Oxidative Phosphorylation in Human Brain Tissue. Bipolar Disord. 2009, 11, 515–522. [Google Scholar] [CrossRef]
- Bachmann, R.F.; Schloesser, R.J.; Gould, T.D.; Manji, H.K. Mood Stabilizers Target Cellular Plasticity and Resilience Cascades: Implications for the Development of Novel Therapeutics. Mol. Neurobiol. 2005, 32, 173–202. [Google Scholar] [CrossRef] [PubMed]
- Kazuno, A.-A.; Munakata, K.; Kato, N.; Kato, T. Mitochondrial DNA-Dependent Effects of Valproate on Mitochondrial Calcium Levels in Transmitochondrial Cybrids. Int. J. Neuropsychopharmacol. 2008, 11, 71–78. [Google Scholar] [CrossRef]
- Chen, H.; Dzitoyeva, S.; Manev, H. Effect of Valproic Acid on Mitochondrial Epigenetics. Eur. J. Pharmacol. 2012, 690, 51–59. [Google Scholar] [CrossRef]
- Liang, L.; Chen, J.; Xiao, L.; Wang, Q.; Wang, G. Mitochondrial Modulators in the Treatment of Bipolar Depression: A Systematic Review and Meta-Analysis. Transl. Psychiatry 2022, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Nierenberg, A.A.; Kansky, C.; Brennan, B.P.; Shelton, R.C.; Perlis, R.; Iosifescu, D. V Mitochondrial Modulators for Bipolar Disorder: A Pathophysiologically Informed Paradigm for New Drug Development. Aust. N. Z. J. Psychiatry 2013, 47, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Dean, O.M.; Turner, A.; Malhi, G.S.; Ng, C.; Cotton, S.M.; Dodd, S.; Sarris, J.; Samuni, Y.; Tanious, M.; Dowling, N.; et al. Design and Rationale of a 16-Week Adjunctive Randomized Placebo-Controlled Trial of Mitochondrial Agents for the Treatment of Bipolar Depression. Braz. J. Psychiatry 2015, 37, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, J.D.; Kakar, R.; Berk, M.; Kessing, L.V.; Vinberg, M.; Baune, B.T.; Mansur, R.B.; Brietzke, E.; Goldstein, B.I.; Mcintyre, R.S. Anti-Inflammatory Agents in the Treatment of Bipolar Depression: A Systematic Review and Meta-Analysis. Bipolar Disord. 2016, 18, 89–101. [Google Scholar] [CrossRef]
- Kishi, T.; Miyake, N.; Okuya, M.; Sakuma, K.; Iwata, N. N-Acetylcysteine as an Adjunctive Treatment for Bipolar Depression and Major Depressive Disorder: A Systematic Review and Meta-Analysis of Double-Blind, Randomized Placebo-Controlled Trials. Psychopharmacology 2020, 237, 3481–3487. [Google Scholar] [CrossRef] [PubMed]
- Sarris, J.; Murphy, J.; Mischoulon, D.; Papakostas, G.I.; Fava, M.; Berk, M.; Ng, C.H. Adjunctive Nutraceuticals for Depression: A Systematic Review and Meta-Analyses. Am. J. Psychiatry 2016, 173, 575–587. [Google Scholar] [CrossRef]
- Ashton, M.M.; Kavanagh, B.E.; Marx, W.; Berk, M.; Sarris, J.; Ng, C.H.; Hopwood, M.; Williams, L.J.; Dean, O.M. A Systematic Review of Nutraceuticals for the Treatment of Bipolar Disorder. Can. J. Psychiatry 2021, 66, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Olagunju, A.T.; Morgan, J.A.; Aftab, A.; Gatchel, J.R.; Chen, P.; Dols, A.; Sajatovic, M.; Regenold, W.T. A Review of the Evidence Base for Nutrition and Nutritional Supplements in Older Adults with Bipolar Disorder: A Report from the OABD Task Force. J. Frailty Aging 2021, 10, 241–246. [Google Scholar] [CrossRef]
- Chmiel, I. Ketogenic Diet in Therapy of Bipolar Affective Disorder—Case Report and Literature Review. Psychiatr. Pol. 2022, 56, 1345–1363. [Google Scholar] [CrossRef]
- Seo, J.H.; Park, H.S.; Park, S.S.; Kim, C.J.; Kim, D.H.; Kim, T.W. Physical Exercise Ameliorates Psychiatric Disorders and Cognitive Dysfunctions by Hippocampal Mitochondrial Function and Neuroplasticity in Post-Traumatic Stress Disorder. Exp. Neurol. 2019, 322, 113043. [Google Scholar] [CrossRef]
- Sun, L.; Liu, T.; Liu, J.; Gao, C.; Zhang, X. Physical Exercise and Mitochondrial Function: New Therapeutic Interventions for Psychiatric and Neurodegenerative Disorders. Front. Neurol. 2022, 13, 929781. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Callio, J.; Distefano, G.; O’Doherty, R.M.; Goodpaster, B.H.; Coen, P.M.; Chu, C.T. Exercise Increases Mitochondrial Complex I Activity and DRP1 Expression in the Brains of Aged Mice. Exp. Gerontol. 2017, 90, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.M.; Nolan, Y.M. Neuroinflammation Negatively Affects Adult Hippocampal Neurogenesis and Cognition: Can Exercise Compensate? Neurosci. Biobehav. Rev. 2016, 61, 121–131. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giménez-Palomo, A.; Andreu, H.; de Juan, O.; Olivier, L.; Ochandiano, I.; Ilzarbe, L.; Valentí, M.; Stoppa, A.; Llach, C.-D.; Pacenza, G.; et al. Mitochondrial Dysfunction as a Biomarker of Illness State in Bipolar Disorder: A Critical Review. Brain Sci. 2024, 14, 1199. https://doi.org/10.3390/brainsci14121199
Giménez-Palomo A, Andreu H, de Juan O, Olivier L, Ochandiano I, Ilzarbe L, Valentí M, Stoppa A, Llach C-D, Pacenza G, et al. Mitochondrial Dysfunction as a Biomarker of Illness State in Bipolar Disorder: A Critical Review. Brain Sciences. 2024; 14(12):1199. https://doi.org/10.3390/brainsci14121199
Chicago/Turabian StyleGiménez-Palomo, Anna, Helena Andreu, Oscar de Juan, Luis Olivier, Iñaki Ochandiano, Lidia Ilzarbe, Marc Valentí, Aldo Stoppa, Cristian-Daniel Llach, Giulio Pacenza, and et al. 2024. "Mitochondrial Dysfunction as a Biomarker of Illness State in Bipolar Disorder: A Critical Review" Brain Sciences 14, no. 12: 1199. https://doi.org/10.3390/brainsci14121199
APA StyleGiménez-Palomo, A., Andreu, H., de Juan, O., Olivier, L., Ochandiano, I., Ilzarbe, L., Valentí, M., Stoppa, A., Llach, C.-D., Pacenza, G., Andreazza, A. C., Berk, M., Vieta, E., & Pacchiarotti, I. (2024). Mitochondrial Dysfunction as a Biomarker of Illness State in Bipolar Disorder: A Critical Review. Brain Sciences, 14(12), 1199. https://doi.org/10.3390/brainsci14121199