
Could Alcohol-Related Cognitive Decline Be the Result of Iron-Induced Neuroinflammation?


{kind=link}
{kind=link}
Abstract
1. Introduction
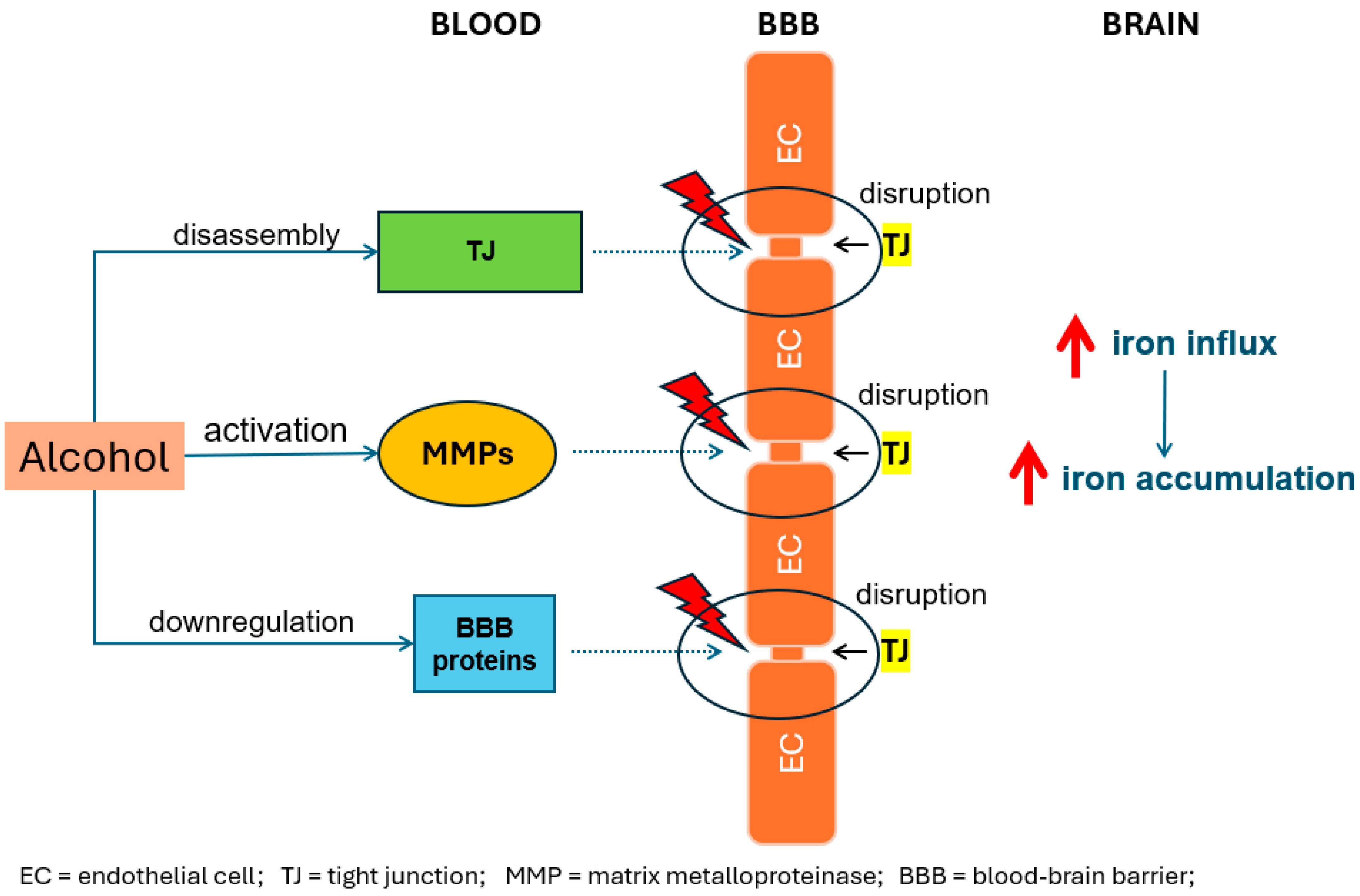
2. Alcohol-Induced Iron Accumulation in the Brain
3. Iron Accumulation in the Brain and Neuroinflammation
4. Neuroinflammation and Cognitive Decline
5. Discussion
6. Conclusions
Funding
Conflicts of Interest
References
- Goodwin, D.W.; Othmer, E.; Halikas, J.A.; Freemon, F. Loss of Short Term Memory as a Predictor of the Alcoholic “Blackout”. Nature 1970, 227, 201–202. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.; Butters, N.; Ryan, C.; Bayog, R. Cognitive Loss and Recovery in Long-Term Alcohol Abusers. Arch. Gen. Psychiatry 1983, 40, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Topiwala, A.; Allan, C.L.; Valkanova, V.; Zsoldos, E.; Filippini, N.; Sexton, C.; Mahmood, A.; Fooks, P.; Singh-Manoux, A.; Mackay, C.E.; et al. Moderate Alcohol Consumption as Risk Factor for Adverse Brain Outcomes and Cognitive Decline: Longitudinal Cohort Study. BMJ 2017, 357, j2353. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef] [PubMed]
- Ilomaki, J.; Jokanovic, N.; Tan, E.C.K.; Lonnroos, E. Alcohol Consumption, Dementia and Cognitive Decline: An Overview of Systematic Reviews. Curr. Clin. Pharmacol. 2015, 10, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Nutt, D.; Hayes, A.; Fonville, L.; Zafar, R.; Palmer, E.O.C.; Paterson, L.; Lingford-Hughes, A. Alcohol and the Brain. Nutrients 2021, 13, 3938. [Google Scholar] [CrossRef] [PubMed]
- Schwarzinger, M.; Pollock, B.G.; Hasan, O.S.M.; Dufouil, C.; Rehm, J.; QalyDays Study Group. Contribution of Alcohol Use Disorders to the Burden of Dementia in France 2008-13: A Nationwide Retrospective Cohort Study. Lancet Public Health 2018, 3, e124–e132. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, H.; Wan, Y.; Tan, C.; Li, J.; Tan, L.; Yu, J.-T. Alcohol Consumption and Dementia Risk: A Dose-Response Meta-Analysis of Prospective Studies. Eur. J. Epidemiol. 2017, 32, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Piumatti, G.; Moore, S.; Berridge, D.; Sarkar, C.; Gallacher, J. The Relationship between Alcohol Use and Long-Term Cognitive Decline in Middle and Late Life: A Longitudinal Analysis Using UK Biobank. J. Public Health 2018, 40, 313–314. [Google Scholar] [CrossRef]
- Sabia, S.; Fayosse, A.; Dumurgier, J.; Dugravot, A.; Akbaraly, T.; Britton, A.; Kivimäki, M.; Singh-Manoux, A. Alcohol Consumption and Risk of Dementia: 23 Year Follow-Up of Whitehall II Cohort Study. BMJ 2018, 362, k2927. [Google Scholar] [CrossRef]
- Phillis, J.W.; Jhamandas, K. The Effects of Chlorpromazine and Ethanol on in Vivo Release of Acetylcholine from the Cerebral Cortex. Comp. Gen. Pharmacol. 1971, 2, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Hood, W.F. Inhibition of Synaptosomal Calcium Uptake by Ethanol. J. Pharmacol. Exp. Ther. 1980, 213, 562–568. [Google Scholar] [PubMed]
- Lovinger, D.M.; White, G.; Weight, F.F. Ethanol Inhibits NMDA-Activated Ion Current in Hippocampal Neurons. Science 1989, 243, 1721–1724. [Google Scholar] [CrossRef] [PubMed]
- Reichenberg, A.; Yirmiya, R.; Schuld, A.; Kraus, T.; Haack, M.; Morag, A.; Pollmächer, T. Cytokine-Associated Emotional and Cognitive Disturbances in Humans. Arch. Gen. Psychiatry 2001, 58, 445. [Google Scholar] [CrossRef] [PubMed]
- Montecinos, L.; Eskew, J.D.; Smith, A. What Is next in This “Age” of Heme-Driven Pathology and Protection by Hemopexin? An Update and Links with Iron. Pharmaceuticals 2019, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Ureña-Peralta, J.; Morillo-Bargues, M.J.; Gómez-Pinedo, U.; Guerri, C. Ethanol-Induced TLR4/NLRP3 Neuroinflammatory Response in Microglial Cells Promotes Leukocyte Infiltration Across the BBB. Neurochem. Res. 2016, 41, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Topiwala, A.; Wang, C.; Ebmeier, K.P.; Burgess, S.; Bell, S.; Levey, D.F.; Zhou, H.; McCracken, C.; Roca-Fernández, A.; Petersen, S.E.; et al. Associations between Moderate Alcohol Consumption, Brain Iron, and Cognition in UK Biobank Participants: Observational and Mendelian Randomization Analyses. PLoS Med. 2022, 19, e1004039. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Iron, Neuroinflammation and Neurodegeneration. IJMS 2022, 23, 7267. [Google Scholar] [CrossRef] [PubMed]
- Conway, D.; Henderson, M.A. Iron Metabolism. Anaesth. Intensive Care Med. 2022, 23, 123–125. [Google Scholar] [CrossRef]
- Harrison-Findik, D.D. Role of Alcohol in the Regulation of Iron Metabolism. World J. Gastroenterol. 2007, 13, 4925–4930. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of Iron and Hydrogen Peroxide: The Fenton Reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M. Iron, Free Radicals, and Oxidative Injury. Semin. Hematol. 1998, 35, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Juhás, M.; Sun, H.; Brown, M.R.G.; MacKay, M.B.; Mann, K.F.; Sommer, W.H.; Wilman, A.H.; Dursun, S.M.; Greenshaw, A.J. Deep Grey Matter Iron Accumulation in Alcohol Use Disorder. Neuroimage 2017, 148, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, J.; Shen, Y.; Li, H.; Rausch, W.-D.; Huang, X. Iron Dyshomeostasis and Ferroptosis: A New Alzheimer’s Disease Hypothesis? Front. Aging Neurosci. 2022, 14, 830569. [Google Scholar] [CrossRef]
- Duane, P.; Raja, K.B.; Simpson, R.J.; Peters, T.J. Intestinal Iron Absorption in Chronic Alcoholics. Alcohol Alcohol. 1992, 27, 539–544. [Google Scholar] [PubMed]
- Urrutia, P.J.; Bórquez, D.A.; Núñez, M.T. Inflaming the Brain with Iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef]
- Haorah, J.; Heilman, D.; Knipe, B.; Chrastil, J.; Leibhart, J.; Ghorpade, A.; Miller, D.W.; Persidsky, Y. Ethanol-Induced Activation of Myosin Light Chain Kinase Leads to Dysfunction of Tight Junctions and Blood-Brain Barrier Compromise. Alcohol. Clin. Exp. Res. 2005, 29, 999–1009. [Google Scholar] [CrossRef]
- Rubio-Araiz, A.; Porcu, F.; Pérez-Hernández, M.; García-Gutiérrez, M.S.; Aracil-Fernández, M.A.; Gutierrez-López, M.D.; Guerri, C.; Manzanares, J.; O’Shea, E.; Colado, M.I. Disruption of Blood-Brain Barrier Integrity in Postmortem Alcoholic Brain: Preclinical Evidence of TLR4 Involvement from a Binge-like Drinking Model. Addict. Biol. 2017, 22, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Zima, T.; Fialová, L.; Mestek, O.; Janebová, M.; Crkovská, J.; Malbohan, I.; Stípek, S.; Mikulíková, L.; Popov, P. Oxidative Stress, Metabolism of Ethanol and Alcohol-Related Diseases. J. Biomed. Sci. 2001, 8, 59–70. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of Pro-Inflammatory Cytokines Released from Microglia in Alzheimer’s Disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, M.; Dranka, B.; Kumar, S.N.; Myers, C.R.; Bennett, B.; Garces, A.M.; Dias Duarte Machado, L.G.; Thiebaut, D.; Ouari, O.; et al. Detection of Mitochondria-Generated Reactive Oxygen Species in Cells Using Multiple Probes and Methods: Potentials, Pitfalls, and the Future. J. Biol. Chem. 2018, 293, 10363–10380. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Alcohol, Oxidative Stress, and Free Radical Damage. Alcohol. Res. Health 2003, 27, 277–284. [Google Scholar]
- Yang, Q.-Q.; Zhou, J.-W. Neuroinflammation in the Central Nervous System: Symphony of Glial Cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef] [PubMed]
- Listabarth, S.; König, D.; Vyssoki, B.; Hametner, S. Does Thiamine Protect the Brain from Iron Overload and Alcohol-related Dementia? Alzheimer’s Dement. 2020, 16, 1591–1595. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef] [PubMed]
- Kupershmidt, L.; Youdim, M.B.H. The Neuroprotective Activities of the Novel Multi-Target Iron-Chelators in Models of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis and Aging. Cells 2023, 12, 763. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Nilsson, L.-G. Memory Function in Normal Aging: Memory Function in Normal Aging. Acta Neurol. Scand. 2003, 107, 7–13. [Google Scholar] [CrossRef]
- Tideman, P.; Stomrud, E.; Leuzy, A.; Mattsson-Carlgren, N.; Palmqvist, S.; Hansson, O.; Alzheimer’s Disease Neuroimaging Initiative. Association of β-Amyloid Accumulation with Executive Function in Adults with Unimpaired Cognition. Neurology 2022, 98, e1525–e1533. [Google Scholar] [CrossRef] [PubMed]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in Mild Cognitive Impairment and Alzheimer’s Disease: A Meta-Analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chi, G.C.; Fitzpatrick, A.L.; Sharma, M.; Jenny, N.S.; Lopez, O.L.; DeKosky, S.T. Inflammatory Biomarkers Predict Domain-Specific Cognitive Decline in Older Adults. Gerona 2016, glw155. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Panda, S.R.; Kwatra, M.; Sahu, B.D.; Naidu, V. Perillyl Alcohol Attenuates NLRP3 Inflammasome Activation and Rescues Dopaminergic Neurons in Experimental In Vitro and In Vivo Models of Parkinson’s Disease. ACS Chem. Neurosci. 2022, 13, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Yen, F.-S.; Wang, S.-I.; Lin, S.-Y.; Chao, Y.-H.; Wei, J.C.-C. The Impact of Heavy Alcohol Consumption on Cognitive Impairment in Young Old and Middle Old Persons. J. Transl. Med. 2022, 20, 155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, M.; Chang, W. Iron Dyshomeostasis and Ferroptosis in Alzheimer’s Disease: Molecular Mechanisms of Cell Death and Novel Therapeutic Drugs and Targets for AD. Front. Pharmacol. 2022, 13, 983623. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s Disease: Pathogenesis, Diagnostics, and Therapeutics. IJN 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Quispe, C.; Castillo, C.M.S.; Caroca, R.; Lazo-Vélez, M.A.; Antonyak, H.; Polishchuk, A.; Lysiuk, R.; Oliinyk, P.; De Masi, L.; et al. Ellagic Acid: A Review on Its Natural Sources, Chemical Stability, and Therapeutic Potential. Oxidative Med. Cell. Longev. 2022, 2022, 3848084. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma Frequency Entrainment Attenuates Amyloid Load and Modifies Microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef]
- Parhizkar, S.; Holtzman, D.M. APOE Mediated Neuroinflammation and Neurodegeneration in Alzheimer’s Disease. Semin. Immunol. 2022, 59, 101594. [Google Scholar] [CrossRef]
- Kagerer, S.M.; Schroeder, C.; van Bergen, J.M.G.; Schreiner, S.J.; Meyer, R.; Steininger, S.C.; Vionnet, L.; Gietl, A.F.; Treyer, V.; Buck, A.; et al. Low Subicular Volume as an Indicator of Dementia-Risk Susceptibility in Old Age. Front. Aging Neurosci. 2022, 14, 811146. [Google Scholar] [CrossRef] [PubMed]
- Mehndiratta, P.; Manjila, S.; Ostergard, T.; Eisele, S.; Cohen, M.L.; Sila, C.; Selman, W.R. Cerebral Amyloid Angiopathy–Associated Intracerebral Hemorrhage: Pathology and Management. FOC 2012, 32, E7. [Google Scholar] [CrossRef] [PubMed]
- Carreno, G.; Guiho, R.; Martinez-Barbera, J.P. Cell Senescence in Neuropathology: A Focus on Neurodegeneration and Tumours. Neuropathol. Appl. Neurobiol. 2021, 47, 359–378. [Google Scholar] [CrossRef]
- Rodríguez-González, A.; Moya, M.; Rodríguez De Fonseca, F.; Gómez De Heras, R.; Orio, L. Alcohol Binge Drinking Induces Downregulation of Blood-Brain Barrier Proteins in the Rat Frontal Cortex -but Not in the Hippocampus-That Is Not Prevented by OEA Pretreatment. Adv. Drug Alcohol. Res. 2023, 3, 11091. [Google Scholar] [CrossRef]
- Pratt, O.E.; Rooprai, H.K.; Shaw, G.K.; Thomson, A.D. The genesis of alcoholic brain tissue injury. Alcohol Alcohol. 1990, 25, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Schall, K.; Ramirez, S.H.; Persidsky, Y. Activation of Protein Tyrosine Kinases and Matrix Metalloproteinases Causes Blood-brain Barrier Injury: Novel Mechanism for Neurodegeneration Associated with Alcohol Abuse. Glia 2008, 56, 78–88. [Google Scholar] [CrossRef]
- Kopelman, M.D. Frontal Dysfunction and Memory Deficits in the Alcoholic Korsakoff Syndrome and Alzheimer-Type Dementia. Brain 1991, 114 Pt 1A, 117–137. [Google Scholar]
- Shi, Z.; El-Obeid, T.; Li, M.; Xu, X.; Liu, J. Iron-Related Dietary Pattern Increases the Risk of Poor Cognition. Nutr. J. 2019, 18, 48. [Google Scholar] [CrossRef]
- Van Bergen, J.M.G.; Li, X.; Hua, J.; Schreiner, S.J.; Steininger, S.C.; Quevenco, F.C.; Wyss, M.; Gietl, A.F.; Treyer, V.; Leh, S.E.; et al. Colocalization of Cerebral Iron with Amyloid Beta in Mild Cognitive Impairment. Sci. Rep. 2016, 6, 35514. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Reichert, C.O.; De Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.D.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. IJMS 2020, 21, 8765. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fu, J.; Zhao, Y.; Liu, Q.; Yan, X.; Su, J. Iron and Targeted Iron Therapy in Alzheimer’s Disease. IJMS 2023, 24, 16353. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, P.; Zhong, M.-L.; Wang, T.; Huang, X.-S.; Li, J.-Y.; Wang, Z.-Y. Deferoxamine Inhibits Iron Induced Hippocampal Tau Phosphorylation in the Alzheimer Transgenic Mouse Brain. Neurochem. Int. 2013, 62, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, O.D.; Schreiner, T.G. Iron Chelators as a Therapeutic Option for Alzheimer’s Disease—A Mini-Review. Front. Aging 2023, 4, 1234958. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Wang, D.-W.; Xu, S.-F.; Zhang, S.; Fan, Y.-G.; Yang, Y.-Y.; Guo, S.-Q.; Wang, S.; Guo, T.; Wang, Z.-Y.; et al. α-Lipoic Acid Improves Abnormal Behavior by Mitigation of Oxidative Stress, Inflammation, Ferroptosis, and Tauopathy in P301S Tau Transgenic Mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilcockson, T.D.W.; Roy, S. Could Alcohol-Related Cognitive Decline Be the Result of Iron-Induced Neuroinflammation? Brain Sci. 2024, 14, 520. https://doi.org/10.3390/brainsci14060520
Wilcockson TDW, Roy S. Could Alcohol-Related Cognitive Decline Be the Result of Iron-Induced Neuroinflammation? Brain Sciences. 2024; 14(6):520. https://doi.org/10.3390/brainsci14060520
Chicago/Turabian StyleWilcockson, Thomas D. W., and Sankanika Roy. 2024. "Could Alcohol-Related Cognitive Decline Be the Result of Iron-Induced Neuroinflammation?" Brain Sciences 14, no. 6: 520. https://doi.org/10.3390/brainsci14060520
APA StyleWilcockson, T. D. W., & Roy, S. (2024). Could Alcohol-Related Cognitive Decline Be the Result of Iron-Induced Neuroinflammation? Brain Sciences, 14(6), 520. https://doi.org/10.3390/brainsci14060520