An In Vivo and In Silico Approach Reveals Possible Sodium Channel Nav1.2 Inhibitors from Ficus religiosa as a Novel Treatment for Epilepsy



Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Plant Material, Extract Preparation, and Fractionation
2.2. Experimental Animal Studies
2.2.1. In Vivo Study Design
2.2.2. Maximal Electroshock (MES)-Induced Seizure Test
2.2.3. Statistical Analysis
2.3. In Silico Studies
2.3.1. Preparation of Protein
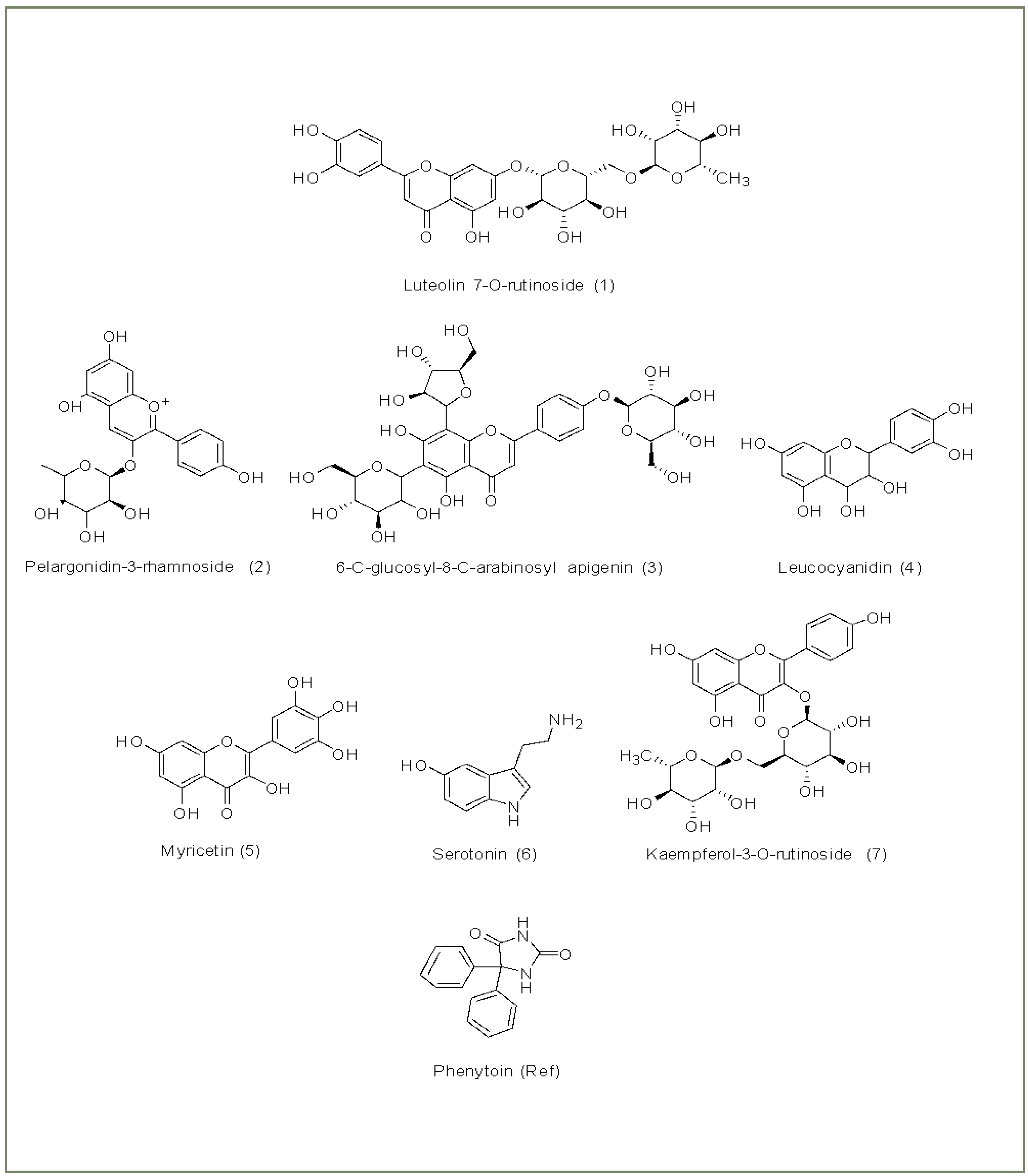
2.3.2. Ligand Selection and Preparation
2.3.3. Molecular Docking
2.3.4. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) Simulation
2.3.5. Molecular Dynamics (MD) Simulation
2.3.6. Absorption, Distribution, Metabolism, Elimination (ADME) Analysis
2.3.7. Toxicity Prediction Study
3. Results
3.1. Maximal Electroshock (MES)-Induced Seizure Model
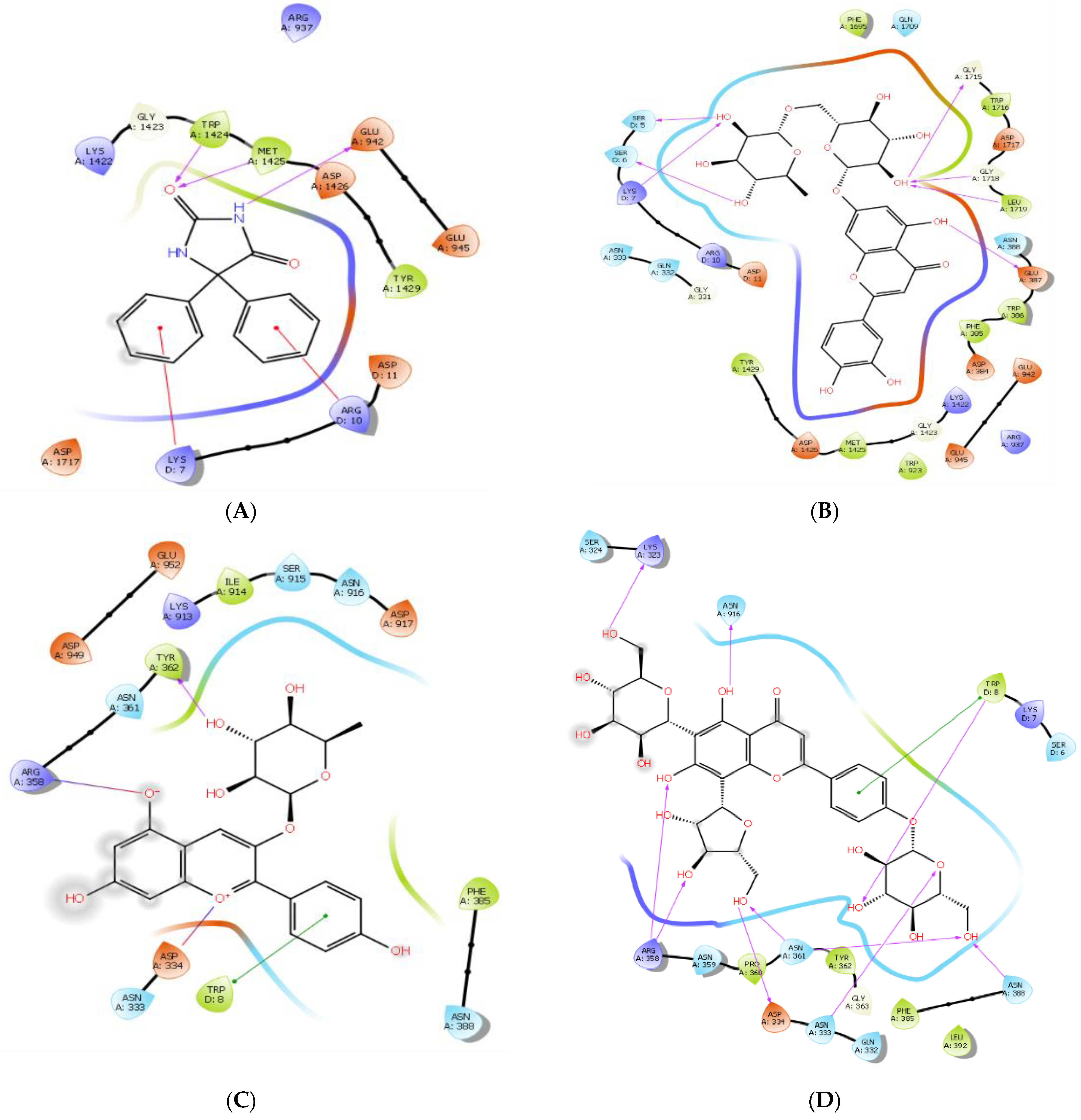
3.2. Molecular Docking
3.3. MM/GBSA Studies
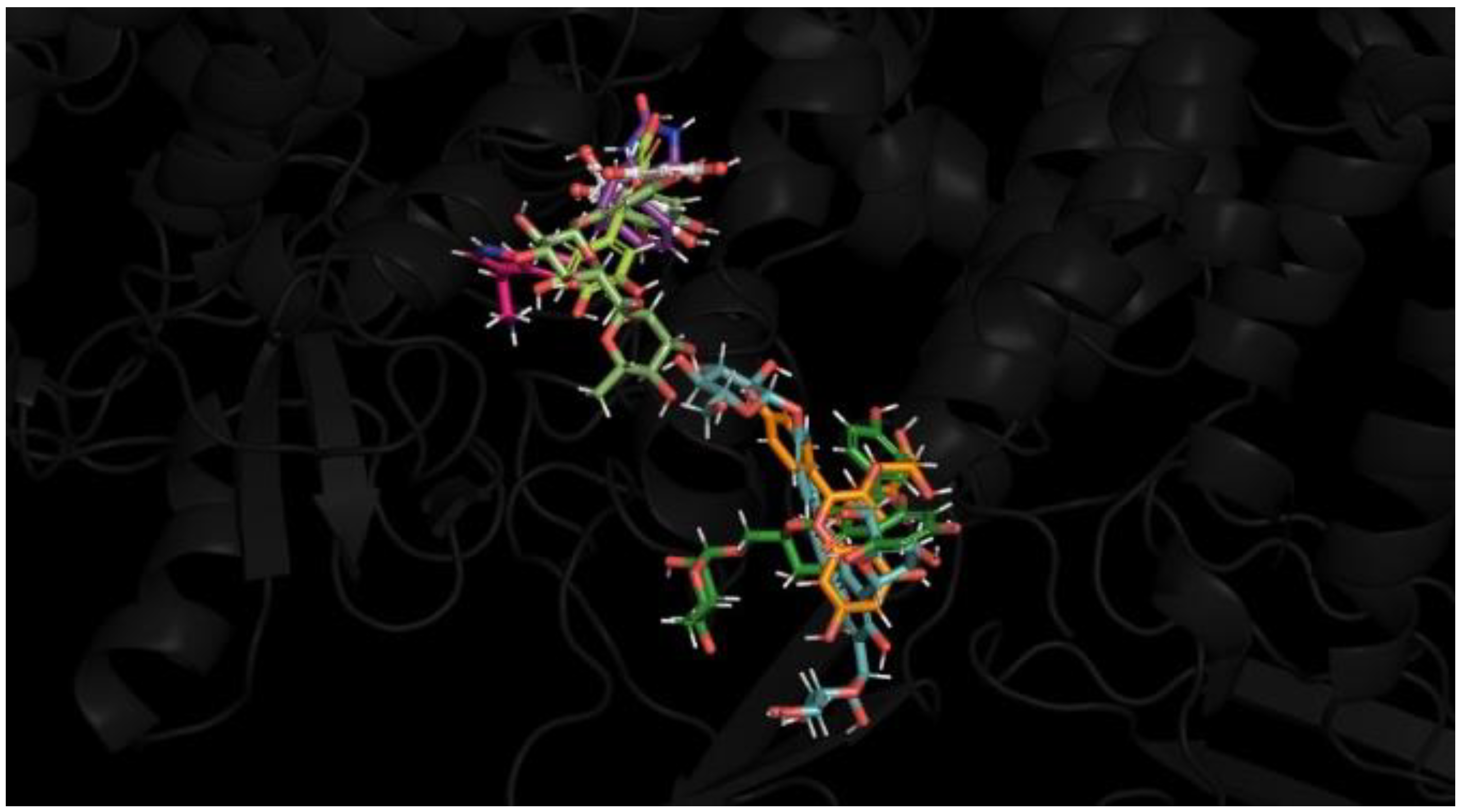
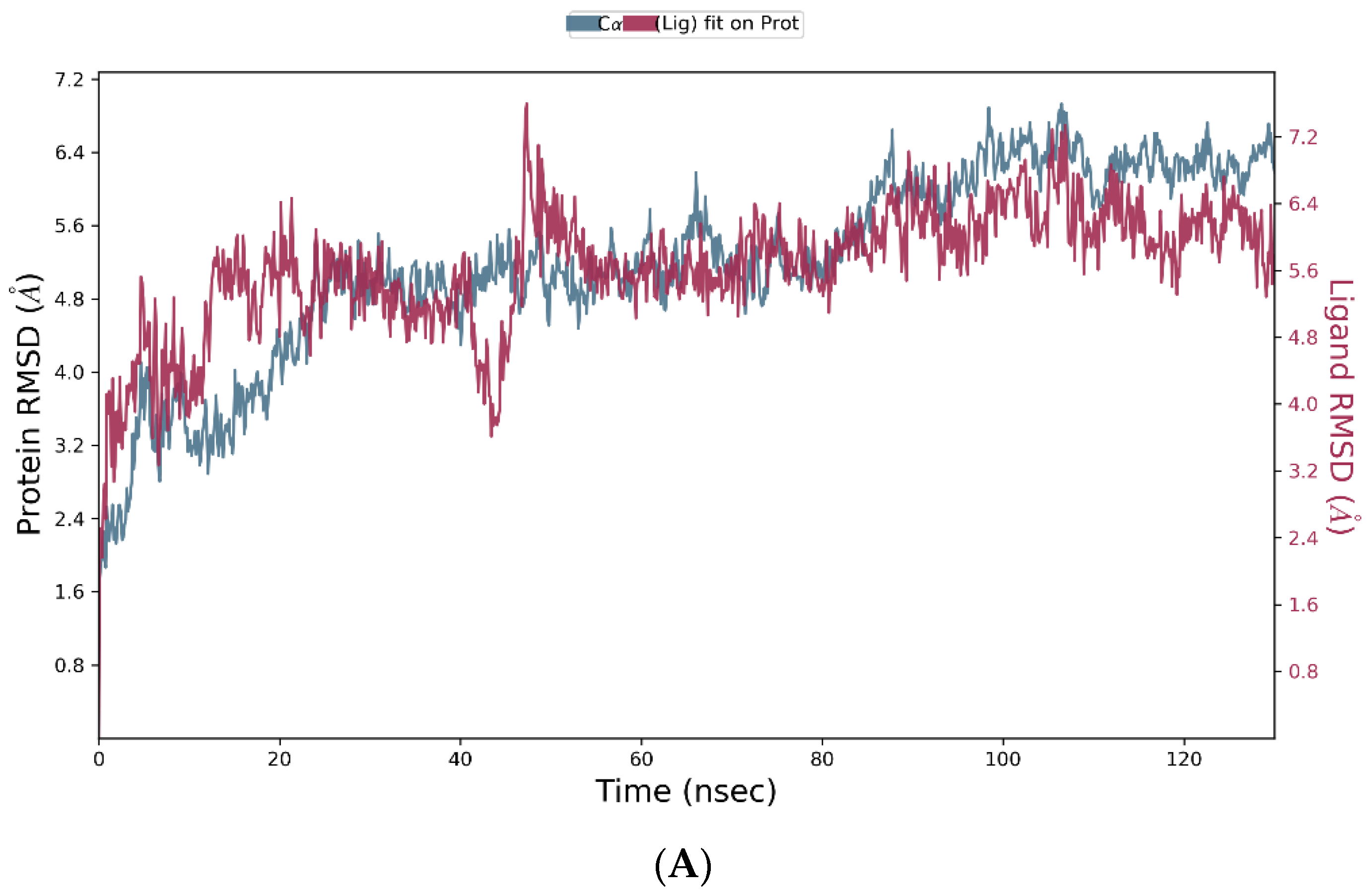
3.4. Molecular Dynamics Simulation
3.5. Drug-likeness Predictions
3.6. Toxicity Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic seizures and epilepsy: Definitions proposed by the International League against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Camerino, D.C.; Tricarico, D. Major channels involved in neuropsychiatric disorders and therapeutic perspectives. Front. Genet. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, U.; Akka, J.; Hinore, J.S.; Girdhar, A.; Bandaru, S.; Sumithnath, T.G.; Nayarisseri, A.; Munshi, A. Computer aided identification of sodium channel blockers in the clinical treatment of epilepsy using molecular docking tools. Bioinformation 2015, 11, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Chanda, B. Sodium channels caught in the act. Science 2019, 363, 1278–1279. [Google Scholar] [CrossRef] [PubMed]
- Ademuwagun, I.A.; Rotimi, S.O.; Syrbe, S.; Ajamma, Y.U.; Adebiyi, E. Voltage Gated Sodium Channel Genes in Epilepsy: Mutations, Functional Studies, and Treatment Dimensions. Front. Neurol. 2021, 12, 600050. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; Chapman, M.L.; Marron, B.E.; Prime, R.; Storer, R.I.; Swain, N.A. Recent progress in sodium channel modulators for pain. Bioorg. Med. Chem. Lett. 2014, 24, 3690–3699. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, M.; Curia, G.; Biagini, G.; Ragsdale, D.S.; Avoli, M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010, 9, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.F.; Sancheti, J.; Sathaye, S. Effect of Eclipta alba on Acute Seizure Models: A GABAA-mediated Effect. Indian J. Pharm. Sci. 2013, 75, 380–384. [Google Scholar] [PubMed]
- Schmidt, D. Drug treatment of epilepsy: Options and limitations. Epilepsy Behav. 2009, 15, 56–65. [Google Scholar] [CrossRef]
- Elger, C.E.; Schmidt, D. Modern management of epilepsy: A practical approach. Epilepsy Behav. 2008, 12, 501–539. [Google Scholar] [CrossRef]
- Zaccara, G.; Franciotta, D.; Perucca, E. Idiosyncratic adverse reactions to antiepileptic drugs. Epilepsia 2007, 48, 1223–1244. [Google Scholar] [CrossRef] [PubMed]
- Kluger, B.M.; Meador, K.J. Teratogenicity of antiepileptic medications. Semin. Neurol. 2008, 28, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Ayalew, M.B.; Muche, E.A. Patient reported adverse events among epileptic patients taking antiepileptic drugs. SAGE Open Med. 2018, 6, 2050312118772471. [Google Scholar] [CrossRef]
- Sultana, B.; Panzini, M.A.; Veilleux Carpentier, A.; Comtois, J.; Rioux, B.; Gore, G.; Bauer, P.R.; Kwon, C.S.; Jetté, N.; Josephson, C.B.; et al. Incidence and Prevalence of Drug-Resistant Epilepsy: A Systematic Review and Meta-analysis. Neurology 2021, 96, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Dell’Isola, G.B.; Vinti, V.; Fattorusso, A.; Tascini, G.; Mencaroni, E.; Di Cara, G.; Striano, P.; Verrotti, A. The Broad Clinical Spectrum of Epilepsies Associated with Protocadherin 19 Gene Mutation. Front. Neurol. 2022, 12, 780053. [Google Scholar] [CrossRef] [PubMed]
- Agbo, J.; Ibrahim, Z.G.; Magaji, S.Y.; Mutalub, Y.B.; Mshelia, P.P.; Mhyha, D.H. Therapeutic efficacy of voltage-gated sodium channel inhibitors in epilepsy. Acta Epileptol. 2023, 5, 16. [Google Scholar] [CrossRef]
- Zeng, Q.; Yang, Y.; Duan, J.; Niu, X.; Chen, Y.; Wang, D.; Zhang, J.; Chen, J.; Yang, X.; Li, J.; et al. SCN2A-Related Epilepsy: The Phenotypic Spectrum, Treatment and Prognosis. Front. Mol. Neurosci. 2022, 15, 809951. [Google Scholar] [CrossRef] [PubMed]
- Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 21 May 2024).
- Kinghorn, A.D.; Pan, L.; Fletcher, J.N.; Chai, H. The relevance of higher plants in lead compound discovery programs. J. Nat. Prod. 2011, 74, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Süntar, I. Importance of ethnopharmacological studies in drug discovery: Role of medicinal plants. Phytochem. Rev. 2020, 19, 1199–1209. [Google Scholar] [CrossRef]
- Tiwari, D.; Shukla, P. An overview of the religious and medicinal tree—Ficus religiosa. World J. Pharm. Res. 2023, 12, 443–457. [Google Scholar]
- Vyawahare, N.S.; Khandelwal, A.R.; Batra, V.R.; Nikam, A. Herbal anticonvulsants. J. Herb. Med. Toxicol. 2007, 1, 9–14. [Google Scholar]
- Singh, D.; Singh, B.; Goel, R.K. Hydroethanolic leaf extract of Ficus religiosa lacks anticonvulsant activity in acute electro and chemo convulsion mice models. J. Pharm. Negat. Results 2011, 2, 58–61. [Google Scholar]
- Singh, D.D.; Goel, R. Anticonvulsant effect of Ficus religiosa: Role of serotonergic pathways. J. Ethnopharmacol. 2009, 123, 330–334. [Google Scholar] [CrossRef]
- Singh, D.; Gawande, D.Y.; Singh, T.; Poroikov, V.; Goel, R.K. Revealing pharmacodynamics of medicinal plants using in silico approach: A case study with wet lab validation. Comput. Biol. Med. 2014, 47, 1–6. [Google Scholar] [CrossRef]
- Thakur, D.; Lal, U.R.; Kapoor, D.N.; Kumar, D. Identification and Quantification of Polyphenolic Secondary Metabolites in Stem Bark of Ficus religiosa (Moraceae) Using UPLC-HRMS and RP-HPLC-PDA. Separations 2023, 10, 338. [Google Scholar] [CrossRef]
- Singh, P.; Singh, D.; Goel, R.K. Ficus religiosa, L. figs—A potential herbal adjuvant to phenytoin for improved management of epilepsy and associated behavioral comorbidities. Epilepsy Behav. 2014, 41, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Ratnasooriya, W.D.; Jayakody, J.R.A.C.; Dharmasiri, M.G.J.M.S.R. An Aqueous extract of trunk bark of Ficus Religiosa has anxiolytic activity. Med. Sci. Res. 1998, 26, 817–819. [Google Scholar]
- Bhangale, J.O.; Acharya, S.R. Anti-Parkinson Activity of Petroleum Ether Extract of Ficus religiosa (L.) Leaves. Adv. Pharmacol. Sci. 2016, 2016, 9436106. [Google Scholar]
- Sharma, L.; Mishra, J.; Sharma, D. Pharmacological Role of Ficus Religiosa A Hidden Sacred Fig Plant: A Systematic Literature Review. Eur. Chem. Bull. 2023, 12, 3054–3080. [Google Scholar]
- Balunas, M.J.; Kinghorn, A.D. Drug discovery from medicinal plants. Life Sci. 2005, 78, 431–441. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed]
- Duarte, Y.; Márquez-Miranda, V.; Miossec, M.J.; González-Nilo, F. Integration of target discovery, drug discovery and drug delivery: A review on computational strategies. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1554. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Wilcox, K.S.; White, H.S. Discovery of antiepileptic drugs. Neurotherapeutics 2007, 4, 12–17. [Google Scholar] [CrossRef]
- Kehne, J.H.; Klein, B.D.; Raeissi, S.; Sharma, S. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem. Res. 2017, 42, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Stables, J.P.; Bertram, E.H.; White, H.S.; Coulter, D.A.; Dichter, M.A.; Jacobs, M.P.; Loscher, W.; Lowenstein, D.H.; Moshe, S.L.; Noebels, J.L.; et al. Models for epilepsy and epileptogenesis: Report from the NIH workshop, Bethesda, Maryland. Epilepsia 2002, 43, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Wlaź, P. Acute Seizure Tests Used in Epilepsy Research: Step-by-Step Protocol of the Maximal Electroshock Seizure (MES) Test, the Maximal Electroshock Seizure Threshold (MEST) Test, and the Pentylenetetrazole (PTZ)-Induced Seizure Test in Rodents. In Experimental and Translational Methods to Screen Drugs Effective against Seizures and Epilepsy; Neuromethods; Vohora, D., Ed.; Humana: New York, NY, USA, 2021; Volume 167. [Google Scholar] [CrossRef]
- Löscher, W.; Schmidt, D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res. 1988, 2, 145–181. [Google Scholar] [CrossRef] [PubMed]
- Large, C.H.; Kalinichev, M.; Lucas, A.; Carignani, C.; Bradford, A.; Garbati, N.; Sartori, I.; Austin, N.E.; Ruffo, A.; Jones, D.N.; et al. The relationship between sodium channel inhibition and anticonvulsant activity in a model of generalised seizure in the rat. Epilepsy Res. 2009, 85, 96–106. [Google Scholar] [CrossRef] [PubMed]
- White, H.S. Clinical significance of animal seizure models and mechanism of action studies of potential antiepileptic drugs. Epilepsia 1997, 38 (Suppl. 1), S9–S17. [Google Scholar]
- Patocka, J.; Wu, Q.; Nepovimova, E.; Kuca, K. Phenytoin—An anti-seizure drug: Overview of its chemistry, pharmacology and toxicology. Food Chem. Toxicol. 2020, 142, 111393. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Kamalinejad, M.; Javadi, M.; Majzoob, S.; Sayyah, M. Evaluation of the anticonvulsant activity of the essential oil of Eugenia caryophyllata in male mice. J. Ethnopharmacol. 1999, 64, 167–171. [Google Scholar] [CrossRef]
- Singh, D.; Singh, B.; Goel, R.K. Role of saponins for the anticonvulsant effect of adventitious roots of Ficus religiosa. Pharm. Biol. 2012, 50, 816–822. [Google Scholar] [CrossRef]
- Ajeet, A.; Kumar, A.; Mishra, A. Design, Synthesis and Pharmacological Evaluation of Sulfonamide Derivatives Screened against Maximal Electroshock Seizure Test. Mol. Biol. 2018, 7, 1–10. [Google Scholar] [CrossRef]
- Al-Snafi, A. Pharmacology of Ficus religiosa—A review. IOSR J. Pharm. 2017, 7, 61–71. [Google Scholar] [CrossRef]
- Swami, K.; Bisht, N. Constituents of Ficus religiosa and Ficus infectoria and their biological activity. J. Indian Chem. Soc. 1996, 73, 631. [Google Scholar]
- Murugesu, S.; Selamat, J.; Perumal, V. Phytochemistry, Pharmacological Properties, and Recent Applications of Ficus benghalensis and Ficus religiosa. Plants 2021, 10, 2749. [Google Scholar] [CrossRef] [PubMed]
- Grison-Pigé, L.; Hossaert-McKey, M.; Greeff, J.M.; Bessière, J.M. Fig volatile compounds—A first comparative study. Phytochemistry 2002, 61, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible ligand docking with Glide. Curr. Protoc. Bioinform. 2007, 18, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Salo-Ahen, O.; Alanko, I.; Bhadane, R.; Bonvin, A.M.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2020, 9, 71. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Garg, S.; Anand, A.; Lamba, Y.; Roy, A. Molecular docking analysis of selected phytochemicals against SARS-CoV-2 Mpro receptor. Vegetos 2020, 33, 766–781. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC ’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- David, T.I.; Adelakun, N.S.; Omotuyi, O.I.; Metibemu, D.S.; Ekun, O.E.; Eniafe, G.O.; Inyang, O.K.; Adewumi, B.; Enejoh, O.A.; Owolabi, R.T.; et al. Molecular docking analysis of phyto-constituents from Cannabis sativa with pfDHFR. Bioinformation 2018, 14, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Yunta, M. Docking and Ligand Binding Affinity: Uses and Pitfalls. Am. J. Model. Optim. 2016, 4, 74–114. [Google Scholar]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef]
- Scheiner, S.; Kar, T.; Pattanayak, J. Comparison of various types of hydrogen bonds involving aromatic amino acids. J. Am. Chem. Soc. 2002, 124, 13257–13264. [Google Scholar] [CrossRef]
- Levitt, M.; Perutz, M.F. Aromatic rings act as hydrogen bond acceptors. J. Mol. Biol. 1988, 201, 751–754. [Google Scholar] [CrossRef]
- Zhang, X.; Perez-Sanchez, H.; Lightstone, F.C. A Comprehensive Docking and MM/GBSA Rescoring Study of Ligand Recognition upon Binding Antithrombin. Curr. Top. Med. Chem. 2017, 17, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Elekofehinti, O.O.; Iwaloye, O.; Josiah, S.S.; Lawal, A.O.; Akinjiyan, M.O.; Ariyo, E.O. Molecular docking studies, molecular dynamics and ADME/tox reveal therapeutic potentials of STOCK1N-69160 against papain-like protease of SARS-CoV-2. Mol. Divers. 2021, 25, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Aier, I.; Varadwaj, P.K.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.S.; Patil, C.R.; Patil, S.W.; Jadhav, R.B. Anticonvulsant activity of aqueous root extract of Ficus religiosa. J. Ethnopharmacol. 2011, 133, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Goel, R.K. Anticonvulsant mechanism of saponins fraction from adventitious roots of Ficus religiosa: Possible modulation of GABAergic, calcium and sodium channel functions. Rev. Bras. Farmacogn. 2016, 26, 579–585. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.C. A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Mol. Pharmacol. 1998, 54, 712–721. [Google Scholar]
- Lipkind, G.M.; Fozzard, H.A. Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol. Pharmacol. 2010, 78, 631–638. [Google Scholar] [CrossRef]
- Shih, P.H.; Wu, C.H.; Yeh, C.T.; Yen, G.C. Protective effects of anthocyanins against amyloid β-peptide-induced damage in neuro-2A cells. J. Agric. Food Chem. 2011, 59, 1683–1689. [Google Scholar] [CrossRef]
- Medina dos Santos, N.; Batista, P.B.; Batista, A.G.; Junior, R.M. Current evidence on cognitive improvement and neuroprotection promoted by anthocyanins. Curr. Opin. Food Sci. 2019, 26, 71–78. [Google Scholar] [CrossRef]
- Singh, P.; Singh, D.; Goel, R.K. Phytoflavonoids: Antiepileptics for the future. Int. J. Pharm. Pharm. Sci. 2014, 6, 51–63. [Google Scholar]
- Hirawan, R.; Beta, T. C-Glycosylflavone and Lignan Diglucoside Contents of Commercial, Regular, and Whole-Wheat Spaghetti. Cereal Chem. 2011, 88, 338–343. [Google Scholar] [CrossRef]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The Therapeutic Potential of Apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.M.; Kim, J.; Chung, H.Y.; Choi, J.S. Isolation of luteolin 7-O-rutinoside and esculetin with potential antioxidant activity from the aerial parts of Artemisia montana. Arch. Pharm. Res. 2000, 23, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Cui, Y.; Wu, J.; Shi, L.; Wen, Q.; Yang, S.; Feng, Y. Luteolin-7-O-rutinoside from Pteris cretica, L. var. nervosa attenuates LPS/D-gal-induced acute liver injury by inhibiting PI3K/AKT/AMPK/NF-κB signaling pathway. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2022, 395, 1283–1295. [Google Scholar] [CrossRef]
- Shaikh, M.F.; Tan, K.N.; Borges, K. Anticonvulsant screening of luteolin in four mouse seizure models. Neurosci. Lett. 2013, 550, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Saeed, F.; Hussain, G.; Imran, A.; Mehmood, Z.; Gondal, T.A.; El-Ghorab, A.; Ahmad, I.; Pezzani, R.; Arshad, M.U.; et al. Myricetin: A comprehensive review on its biological potentials. Food Sci. Nutr. 2021, 9, 5854–5868. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.Q.; Meng, F.H.; Tu, L.X.; Sun, L. Myricetin attenuates the severity of seizures and neuroapoptosis in pentylenetetrazole kindled mice by regulating the of BDNF-TrkB signaling pathway and modulating matrix metalloproteinase-9 and GABAA. Exp. Ther. Med. 2019, 17, 3083–3091. [Google Scholar] [CrossRef]
- Bagdy, G.; Kecskemeti, V.; Riba, P.; Jakus, R. Serotonin and epilepsy. J. Neurochem. 2007, 100, 857–873. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Zhang, H. Hepatoprotective effects of kaempferol 3-O-rutinoside and kaempferol 3-O-glucoside from Carthamus tinctorius, L. on CCl4-induced oxidative liver injury in mice. J. Food Drug Anal. 2015, 23, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Khan, M.A.; Ali Zaidi, S.A.; Muhammad, S. In Silico and In Vivo: Evaluating the Therapeutic Potential of Kaempferol, Quercetin, and Catechin to Treat Chronic Epilepsy in a Rat Model. Front. Bioeng. Biotechnol. 2021, 9, 754952. [Google Scholar] [CrossRef] [PubMed]
- Myricetin, Safety Data Sheet. Available online: https://www.sigmaaldrich.com/GB/en/sds/sigma/m6760?userType=anonymous (accessed on 21 May 2024).
- Luteolin 7-rutinoside, Safety Data Sheet. Available online: https://www.sigmaaldrich.com/GB/en/sds/sigma/smb00200?userType=anonymous (accessed on 21 May 2024).
- Leucocyanidin, Safety Data Sheet. Available online: https://file.medchemexpress.com/batch_PDF/HY-119580/Leucocyanidin-SDS-MedChemExpress.pdf (accessed on 21 May 2024).
- Kaempferol-3-O-rutinoside, Safety Data Sheet. Available online: https://www.sigmaaldrich.com/GB/en/sds/sigma/90242?userType=anonymous (accessed on 21 May 2024).
- Serotonin Hydrochloride, Safety Data Sheet. Available online: https://www.sigmaaldrich.com/GB/en/sds/sigma/h9523?userType=anonymous (accessed on 21 May 2024).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Treatment Groups | Pre-Treatment | No. of Animals | Onset Time for THLE (sec) |
|---|---|---|---|---|
| 1 | Control | Normal Saline | 6 | 11.00 ± 2.97 |
| 2 | Standard | Phenytoin | 6 | 1.17 ± 1.33 ** |
| 3 | PE-Fr | PE-Fr | 6 | 2.67 ± 1.96 * |
| 4 | EA-Fr | EA-Fr | 6 | 3.00 ± 1.26 * |
| Prime MM/GBSA | |||||||
|---|---|---|---|---|---|---|---|
| Ligand | Name | Molecular Formula | Molecular Weight (g/mol) | Docking Score | XPGScore | Glide Score | ΔGbind |
| g/mol | kcal/mol | kcal/mol | kcal/mol | kcal/mol | |||
| 1 | Luteolin 7-O-rutinoside | C27H30O15 | 610.5 | −13.476 | −13.476 | −13.476 | −62.13 |
| 2 | Pelargonidin-3- rhamnoside | C21H21O9+ | 417.4 | −8.894 | −10.301 | −10.301 | −45.82 |
| 3 | 6-C-glucosyl-8-C- arabinosyl apigenin | C26H28O14 | 726.2 | −8.147 | −8.191 | −8.191 | −46.82 |
| 4 | Leucocyanidin | C15H14O7 | 306.3 | −7.523 | −7.523 | −7.523 | −24.34 |
| 5 | Myricetin | C15H10O8 | 318.2 | −7.171 | −7.209 | −7.209 | −21.72 |
| 6 | Serotonin | C10H12N2O | 176.2 | −6.963 | −6.963 | −6.963 | −41.45 |
| 7 | Kaempferol-3-O- rutinoside | C27H30O15 | 594.5 | −6.555 | −6.584 | −6.584 | −64.73 |
| Ref | Phenytoin | C15H12N2O2 | 252.3 | −6.660 | −7.022 | −7.022 | −25.80 |
| Interaction Type | Interacting Residues | |
|---|---|---|
| Phenytoin | Polar Non-polar | Glu-942, Trp-1424, Met-1425 Ser-5, Lys-7, Trp-8, Arg-10, Asp-11, His-12, Trp-923, Arg-937, Glu-945, Phe-1421, Lys-1422, Gly-1423, Asp-1426, Tyr-1429, Gly-1715, Asp-1717, Gly-1718 |
| Luteolin 7-O- rutinoside | Polar Non-polar | Ser-5, Ser-6, Lys-7, Asp-11, Glu-387, Gln-1709, Gly-1715, Gly-1718, Leu-1719 Trp-8, Arg-10, His-12, Gly-331, Asn-333, Asn-361, Asp-384, Phe-385, Trp-386, Asn-388, Gln-391, Trp-923, Arg-937, Glu-942, Glu-945, Lys-1422, Gly-1423, Met-1425, Asp-1426, Tyr-1429, Gly-1690, Phe-1695, Trp-1716, Asp-1717, Leu-1720 |
| Pelargonidin-3-rhamnoside | Polar Non-polar | Asn-333, Arg-358, Tyr-362 Ser-6, Trp-8, His-12, Lys-323, Gln-332, Asp-334, Ala-335, Asn-361, Tyr-364, Phe-385, Asn-388, Lys-913, Ile-914, Ser-915, Asn-916, Asp-917, Asp-949, Glu-952 |
| 6-C-glucosyl-8-C-arabinosyl apigenin | Polar Non-polar | Trp-8, Lys-323, Asn-333, Asp-334, Arg-358, Asn-361, Asn-388, Asn-916 Ser-5, Ser-6, Lys-7, Cys-9, Lys-277, Asp-322, Ser-324, His-325, Phe-326, Gly-331, Gln-332, Leu-336, Asn-359, Pro-360, Tyr-362, Gly-363, Arg-379, Phe-385, Leu-392, Ser-915, Asp-917, Glu-945, Trp-948 |
| Leucocyanidin | Polar Non-polar | Arg-10, Asp-11, Asp-384, Phe-385, Lys-1422, Asp-1426, Asp-1717 Lys-7, Trp-8, His-12, Trp-386, Glu-387, Trp-923, Arg-937, Glu-942, Trp-943, Glu-945, Gln-1417, Gly-1423, Trp-1424, Met-1425, Tyr-1429, Ala-1714, Gly-1715, Gly-1718 |
| Myricetin | Polar Non-polar | Ser-5, Glu-942, Gly-1423, Asp-1426 Ser-6, Lys-7, Arg-10, Phe-385, Glu-387, Asn-388, Arg-937, Phe-1421, Lys-1422, Trp-1424, Met-1425, Tyr-1429, Gly-1715, Asp-1717, Gly-1718, Ala-1721 |
| Serotonin | Polar Non-polar | Arg-10, Arg-922, Asp-1433 Asp-11, Ser-13, Arg-14, Cys-15, Cys-16, Phe-1363, His-1365, Met-1374, Val-1401, Met-1425, Asp-1426, Ile-1427, Tyr-1429, Ala-1430, Pro-1441, Lys-1442, Tyr-1443 |
| Kaempferol-3-O- rutinoside | Polar Non-polar | Trp-8, Asp-334, Asn-359, Tyr-362, Asp-917, Asp-949 Cys-1, Cys-2, Cys-9, His-12, Ser-13, Lys-277, Phe-328, Asn-333, Ala-335, Arg-358, Pro-360, Asn-361, Gly-363, Tyr-364, Ile-914, Ser-915, Asn-916, Glu-952 |
| Structure | Ligand | Hydrogen Bonding | Salt Bridges/(π-π)/(π-Cation)/π-Sigma Interactions | Other Hydrophobic Contacts | Van der Waals Interactions |
|---|---|---|---|---|---|
| Ref | Phenytoin | Lys-7, Glu-942, Trp-1424, Met-1425, Asp-1426 | Arg-10 (π-cation) | Tyr-1429 | Asp-11, Glu-945, Lys-1422, Gly-1423, Asp-1426, Asp-1717 |
| 1 | Luteolin 7-O-Rutinoside | Ser-5, Ser-6, Lys-7, Glu-387, Gly-1715, Gly-1718, Leu-1719 | Lys-7 (π-sigma) | Asp-384, Phe-385, Trp-386, Trp-923, Met-1425, Tyr-1429, Phe-1695, Gln-1709, Trp-1716 | Arg-10, Gly-331, Gln-332, Asn-333, Arg-937, Glu-942, Glu-945, Gly-1423, Asp-1426, Tyr-1429 |
| 2 | Pelargonidin-3-Rhamnoside | Ser-5, Glu-330, Tyr-362, Gly-1690, Asp-1692 | Trp-8 (π-π), Asn-333 (π-lone pair), Asp-334 (salt bridge), Arg-358, Tyr-362 (π-sigma) | Leu-329, Leu-336, Phe-385, Leu-392, Arg-395, Ile-914, Val-1689 | - |
| 3 | 6-C-Glucosyl-8-C-Arabinosyl Apigenin | Trp-8, Lys-323, Asn-333, Asp-334, Arg-358, Asn-361, Asn-388, Asn-916 | Trp-8 (π-π T-shaped stacking) | Pro-360, Tyr-362, Phe-385, Leu-392 | Lys-7, Ser-324, Gly-331, Gln-332, Asn-359, Gly-363 |
| 4 | Leucocyanidin | Asp-384, Glu-387, Asp-1426, Asp-1717 | Lys-7 (π-alkyl), Arg-10 (π-cation) | Phe-385, Trp-1424, Met-1425, Tyr-1429 | Gln-1417, Gly-1423 |
| 5 | Myricetin | Gly-1423, Asp-1717 | Lys-7 (π-cation/π -alkyl) | Phe-385, Trp-1424, Met-1425 | Arg-10, Arg-937, Glu-942, Lys-1422, Gly-1718 |
| 6 | Serotonin | Arg-10, Arg-922, Asp-1426 | Asp-1433 (salt bridge) | Cys-16, Met-1374, Val-1401, Tyr-1429, Ala-1430, Pro-1441, Tyr-1443 | Asp-11, Arg-14, His-1365 |
| 7 | Kaempferol-3-O-Rutinoside | Trp-8, Asp-334, Asn-359, Asn-361, Asp-917 | Arg-358 (π-cation) | Cys-1, Cys-2, Phe-328, Pro-360, Tyr-362, Ile-914, Asp-949 | His-12, Ile-914, Asn-916 |
| Ligand | MW (g/mol) | HB Acceptor | HB Donor | Log P | Molar Refractivity | Rule of Five Violations | Bio- Availability Score | GI Absorption | BBB Permeation |
|---|---|---|---|---|---|---|---|---|---|
| Luteolin 7-O- rutinoside | 610.52 | 16 | 10 | 2.81 | 140.52 | 3 | 0.17 | Low | - |
| Pelargonidin-3-rhamnoside | 417.39 | 9 | 6 | −1.61 | 105.11 | 1 | 0.55 | Low | - |
| 6-C-glucosyl-8-C-arabinosyl apigenin | 726.23 | 17 | 11 | 2.16 | 171.97 | 3 | 0.17 | Low | - |
| Leucocyanidin | 306.27 | 7 | 6 | 1.19 | 75.50 | 1 | 0.55 | High | - |
| Myricetin | 318.24 | 8 | 6 | 1.08 | 80.06 | 1 | 0.55 | Low | - |
| Serotonin | 176.21 | 2 | 3 | 1.18 | 52.80 | 0 | 0.55 | High | + |
| Kaempferol-3-O-rutinoside | 594.52 | 15 | 9 | 2.79 | 139.36 | 3 | 0.17 | Low | - |
| Structure Number | Ligand | Predicted LD50 (mg/kg) | Predicted Toxicity Class | Carcinogenicity |
|---|---|---|---|---|
| 1 | Luteolin-7-O-rutinoside | 5000 | 5 | - |
| 2 | Pelargonidin-3-rhamnoside | 5000 | 5 | - |
| 3 | 6-C-glucosyl-8-C- arabinosyl apigenin | 2300 | 5 | - |
| 4 | Leucocyanidin | 2500 | 5 | - |
| 5 | Myricetin | 159 | 3 | - |
| 6 | Serotonin | 2300 | 5 | - |
| 7 | Kaempferol-3-O-rutinoside | 5000 | 5 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, A.; Ahmed, A.; Juffer, A.H.; Carter, W.G. An In Vivo and In Silico Approach Reveals Possible Sodium Channel Nav1.2 Inhibitors from Ficus religiosa as a Novel Treatment for Epilepsy. Brain Sci. 2024, 14, 545. https://doi.org/10.3390/brainsci14060545
Ashraf A, Ahmed A, Juffer AH, Carter WG. An In Vivo and In Silico Approach Reveals Possible Sodium Channel Nav1.2 Inhibitors from Ficus religiosa as a Novel Treatment for Epilepsy. Brain Sciences. 2024; 14(6):545. https://doi.org/10.3390/brainsci14060545
Chicago/Turabian StyleAshraf, Aqsa, Abrar Ahmed, André H. Juffer, and Wayne G. Carter. 2024. "An In Vivo and In Silico Approach Reveals Possible Sodium Channel Nav1.2 Inhibitors from Ficus religiosa as a Novel Treatment for Epilepsy" Brain Sciences 14, no. 6: 545. https://doi.org/10.3390/brainsci14060545
APA StyleAshraf, A., Ahmed, A., Juffer, A. H., & Carter, W. G. (2024). An In Vivo and In Silico Approach Reveals Possible Sodium Channel Nav1.2 Inhibitors from Ficus religiosa as a Novel Treatment for Epilepsy. Brain Sciences, 14(6), 545. https://doi.org/10.3390/brainsci14060545