
Unraveling the Epigenetic Landscape: Insights into Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis


 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Epigenetics
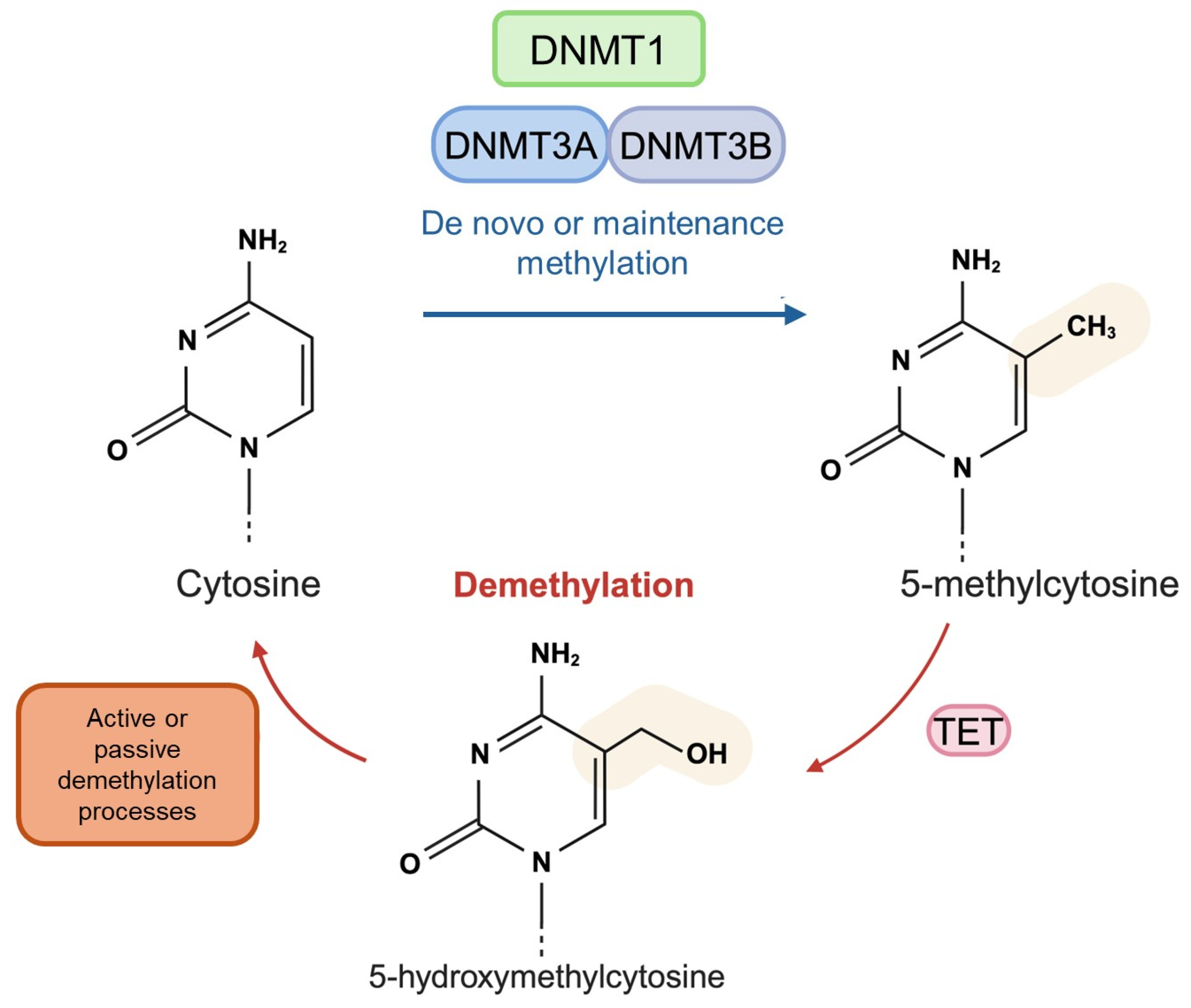
1.2. DNA Methylation and Hydroxymethylation
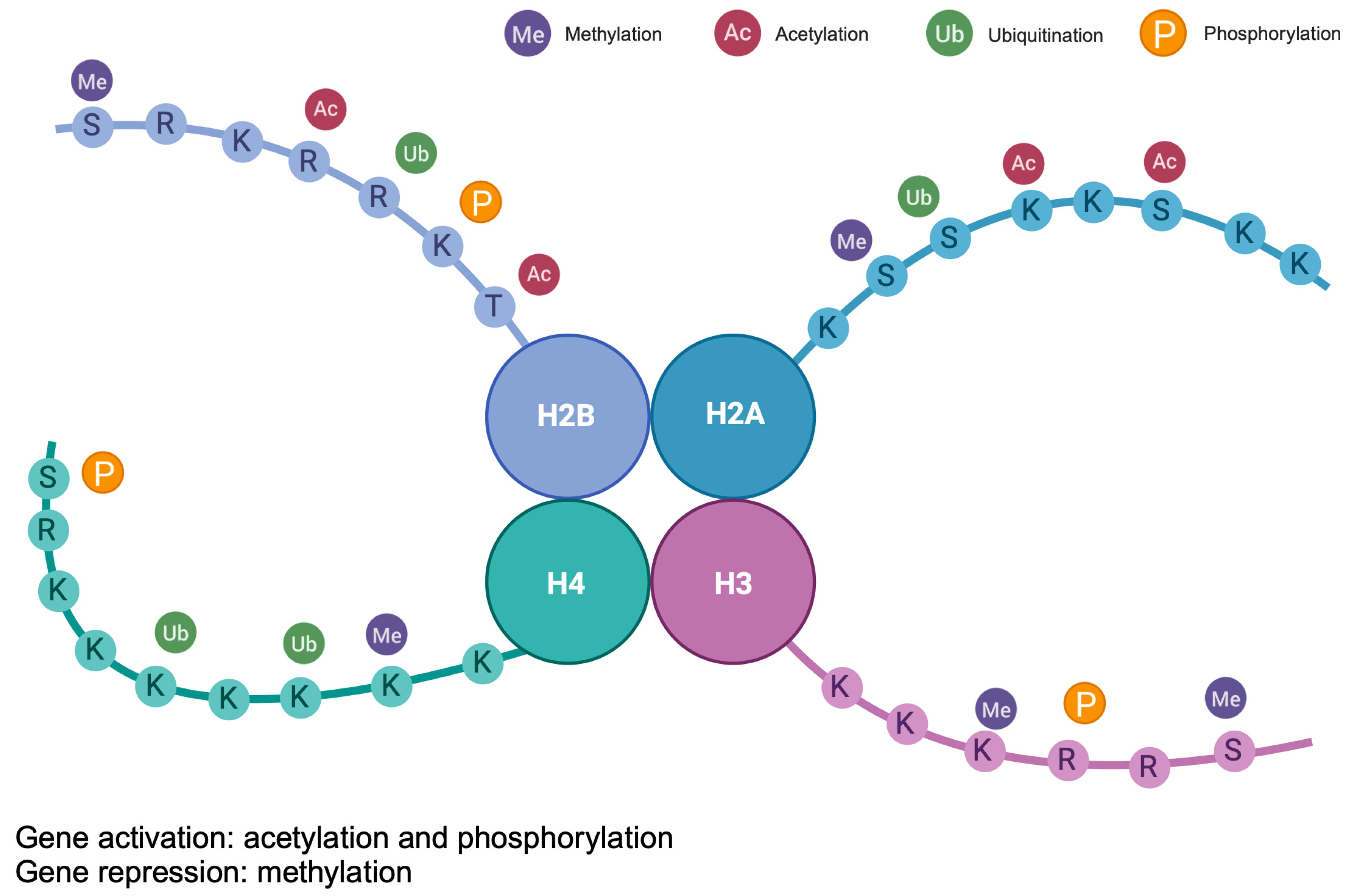
1.3. Histon Modifications
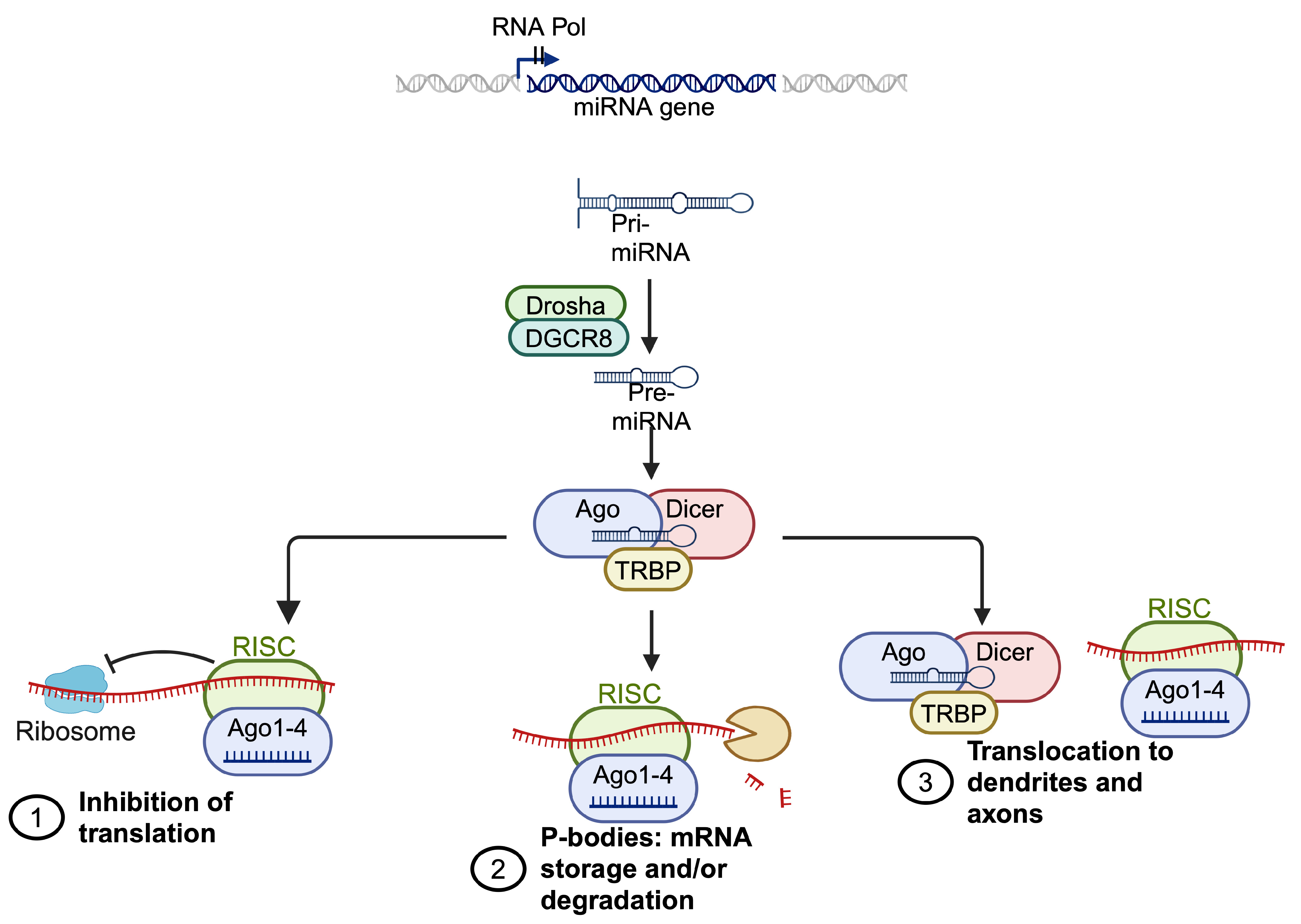
1.4. miRNA Regulation
2. Parkinson’s Disease
2.1. DNA Methylation and Hydroxymethylation in PD
2.2. Histone Posttranslational Modifications in PD
2.3. MicroRNA Regulation in PD
3. Amyotrophic Lateral Sclerosis
3.1. DNA Methylation and Hydroxymethylation in ALS
3.2. Histone Posttranslational Modifications in ALS
3.3. microRNA Regulation in ALS
4. Multiple Sclerosis
4.1. DNA Methylation and Hydroxymethylation in MS
4.2. Histone Modification in MS
4.3. microRNA Regulation in MS
5. Epigenetic Therapy
5.1. Epigenetic Therapy in PD
5.2. Epigenetic Therapy in ALS
5.3. Epigenetic Therapy in MS
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schirinzi, T.; Canevelli, M.; Suppa, A.; Bologna, M.; Marsili, L. The Continuum between Neurodegeneration, Brain Plasticity, and Movement: A Critical Appraisal. Rev. Neurosci. 2020, 31, 723–742. [Google Scholar] [CrossRef]
- Di Credico, A.; Gaggi, G.; Izzicupo, P.; Ferri, L.; Bonanni, L.; Iannetti, G.; Di Baldassarre, A.; Ghinassi, B. Real-Time Monitoring of Levetiracetam Effect on the Electrophysiology of an Heterogenous Human iPSC-Derived Neuronal Cell Culture Using Microelectrode Array Technology. Biosensors 2021, 11, 450. [Google Scholar] [CrossRef]
- Gaggi, G.; Di Credico, A.; Izzicupo, P.; Alviano, F.; Di Mauro, M.; Di Baldassarre, A.; Ghinassi, B. Human Mesenchymal Stromal Cells Unveil an Unexpected Differentiation Potential toward the Dopaminergic Neuronal Lineage. Int. J. Mol. Sci. 2020, 21, 6589. [Google Scholar] [CrossRef]
- Gaggi, G.; Di Credico, A.; Guarnieri, S.; Mariggiò, M.A.; Di Baldassarre, A.; Ghinassi, B. Human Mesenchymal Amniotic Fluid Stem Cells Reveal an Unexpected Neuronal Potential Differentiating into Functional Spinal Motor Neurons. Front. Cell Dev. Biol. 2022, 10, 936990. [Google Scholar] [CrossRef]
- Gaggi, G.; Di Credico, A.; Guarnieri, S.; Mariggiò, M.A.; Ballerini, P.; Di Baldassarre, A.; Ghinassi, B. Human Fetal Membrane-Mesenchymal Stromal Cells Generate Functional Spinal Motor Neurons in Vitro. iScience 2022, 25, 105197. [Google Scholar] [CrossRef]
- Murthy, M.; Cheng, Y.Y.; Holton, J.L.; Bettencourt, C. Neurodegenerative Movement Disorders: An Epigenetics Perspective and Promise for the Future. Neuropathol. Appl. Neurobiol. 2021, 47, 897–909. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Landgrave-Gómez, J.; Mercado-Gómez, O.; Guevara-Guzmán, R. Epigenetic Mechanisms in Neurological and Neurodegenerative Diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [CrossRef]
- Rattan, S.; Flaws, J.A. The Epigenetic Impacts of Endocrine Disruptors on Female Reproduction across Generations. Biol. Reprod. 2019, 101, 635–644. [Google Scholar] [CrossRef]
- Gaggi, G.; Di Credico, A.; Barbagallo, F.; Ballerini, P.; Ghinassi, B.; Di Baldassarre, A. Antenatal Exposure to Plastic Pollutants: Study of the Bisphenols and Perfluoroalkyls Effects on Human Stem Cell Models. Expo. Health 2023. [Google Scholar] [CrossRef]
- Gaggi, G.; Di Credico, A.; Barbagallo, F.; Ghinassi, B.; Di Baldassarre, A. Bisphenols and Perfluoroalkyls Alter Human Stem Cells Integrity: A Possible Link with Infertility. Environ. Res. 2023, 235, 116487. [Google Scholar] [CrossRef] [PubMed]
- Di Credico, A.; Gaggi, G.; Bucci, I.; Ghinassi, B.; Di Baldassarre, A. The Effects of Combined Exposure to Bisphenols and Perfluoroalkyls on Human Perinatal Stem Cells and the Potential Implications for Health Outcomes. Int. J. Mol. Sci. 2023, 24, 15018. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Lin, Z.-J.; Li, C.-C.; Lin, X.; Shan, S.-K.; Guo, B.; Zheng, M.-H.; Li, F.; Yuan, L.-Q.; Li, Z. Epigenetic Regulation in Metabolic Diseases: Mechanisms and Advances in Clinical Study. Signal Transduct. Target. Ther. 2023, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Gaggi, G.; Di Credico, A.; Izzicupo, P.; Antonucci, I.; Crescioli, C.; Di Giacomo, V.; Di Ruscio, A.; Amabile, G.; Alviano, F.; Di Baldassarre, A.; et al. Epigenetic Features of Human Perinatal Stem Cells Redefine Their Stemness Potential. Cells 2020, 9, 1304. [Google Scholar] [CrossRef]
- Borchiellini, M.; Ummarino, S.; Di Ruscio, A. The Bright and Dark Side of DNA Methylation: A Matter of Balance. Cells 2019, 8, 1243. [Google Scholar] [CrossRef]
- Liebl, K.; Zacharias, M. How Methyl–Sugar Interactions Determine DNA Structure and Flexibility. Nucleic Acids Res. 2019, 47, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA Methyltransferase Family: A Versatile Toolkit for Epigenetic Regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Richa, R.; Sinha, R.P. Hydroxymethylation of DNA: An Epigenetic Marker. EXCLI J. 2014, 13, 592–610. [Google Scholar] [PubMed]
- Giallongo, S.; Longhitano, L.; Denaro, S.; D’Aprile, S.; Torrisi, F.; La Spina, E.; Giallongo, C.; Mannino, G.; Lo Furno, D.; Zappalà, A.; et al. The Role of Epigenetics in Neuroinflammatory-Driven Diseases. Int. J. Mol. Sci. 2022, 23, 15218. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET Enzymes in DNA Methylation, Development, and Cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Kozlenkov, A.; Li, J.; Apontes, P.; Hurd, Y.L.; Byne, W.M.; Koonin, E.V.; Wegner, M.; Mukamel, E.A.; Dracheva, S. A Unique Role for DNA (Hydroxy)Methylation in Epigenetic Regulation of Human Inhibitory Neurons. Sci. Adv. 2018, 4, eaau6190. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, E.; Riad, M.; Rao, A.; Heintz, N. 5-Hydroxymethylcytosine-Mediated Active Demethylation Is Required for Mammalian Neuronal Differentiation and Function. eLife 2021, 10, e66973. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Ettig, R.; Kepper, N.; Stehr, R.; Wedemann, G.; Rippe, K. Dissecting DNA-Histone Interactions in the Nucleosome by Molecular Dynamics Simulations of DNA Unwrapping. Biophys. J. 2011, 101, 1999–2008. [Google Scholar] [CrossRef]
- Park, J.; Lee, K.; Kim, K.; Yi, S.-J. The Role of Histone Modifications: From Neurodevelopment to Neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Tiwari, P.; Kaur, A.; Singh, T.G. Sirtuin Acetylation and Deacetylation: A Complex Paradigm in Neurodegenerative Disease. Mol. Neurobiol. 2021, 58, 3903–3917. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gu, Z.; Lin, S.; Chen, L.; Dzreyan, V.; Eid, M.; Demyanenko, S.; He, B. Histone Deacetylases as Epigenetic Targets for Treating Parkinson’s Disease. Brain Sci. 2022, 12, 672. [Google Scholar] [CrossRef] [PubMed]
- Hombach, S.; Kretz, M. Non-Coding RNAs: Classification, Biology and Functioning. In Non-Coding RNAs in Colorectal Cancer; Slaby, O., Calin, G.A., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; Volume 937, pp. 3–17. ISBN 978-3-319-42057-8. [Google Scholar]
- Arab, K.; Park, Y.J.; Lindroth, A.M.; Schäfer, A.; Oakes, C.; Weichenhan, D.; Lukanova, A.; Lundin, E.; Risch, A.; Meister, M.; et al. Long Noncoding RNA TARID Directs Demethylation and Activation of the Tumor Suppressor TCF21 via GADD45A. Mol. Cell 2014, 55, 604–614. [Google Scholar] [CrossRef]
- Zhou, Y.; Yamamoto, Y.; Ochiya, T.; Xiao, Z.; Itaya, T. Distinct Mirna Expression Patterns of Extracellular Vesicles Derived From 4 Types of Mesenchymal Stem Cells. J. Stem Cell Res. Ther. 2018, 8, 415. [Google Scholar] [CrossRef]
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; De Figueiredo Pontes, L.L.; Alberich-Jorda, M.; Zhang, P.; et al. DNMT1-Interacting RNAs Block Gene-Specific DNA Methylation. Nature 2013, 503, 371–376. [Google Scholar] [CrossRef]
- Wilson, R.C.; Doudna, J.A. Molecular Mechanisms of RNA Interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Rubino, E.; Cruciani, M.; Tchitchek, N.; Le Tortorec, A.; Rolland, A.D.; Veli, Ö.; Vallet, L.; Gaggi, G.; Michel, F.; Dejucq-Rainsford, N.; et al. Human Ubiquitin-Specific Peptidase 18 Is Regulated by microRNAs via the 3′Untranslated Region, A Sequence Duplicated in Long Intergenic Non-Coding RNA Genes Residing in Chr22q11.21. Front. Genet. 2021, 11, 627007. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing MiRNA–LncRNA Interactions. In Long Non-Coding RNAs; Feng, Y., Zhang, L., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1402, pp. 271–286. ISBN 978-1-4939-3376-1. [Google Scholar]
- Jankovic, J.; Tan, E.K. Parkinson’s Disease: Etiopathogenesis and Treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Pavlou, M.A.S.; Outeiro, T.F. Epigenetics in Parkinson’s Disease. In Neuroepigenomics in Aging and Disease; Delgado-Morales, R., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 978, pp. 363–390. ISBN 978-3-319-53888-4. [Google Scholar]
- Di Credico, A.; Weiss, A.; Corsini, M.; Gaggi, G.; Ghinassi, B.; Wilbertz, J.H.; Di Baldassarre, A. Machine Learning Identifies Phenotypic Profile Alterations of Human Dopaminergic Neurons Exposed to Bisphenols and Perfluoroalkyls. Sci. Rep. 2023, 13, 21907. [Google Scholar] [CrossRef]
- Papapetropoulos, S.; Glynos, K.; Zhou, Z.; Orfanos, S.E.; Mitsi, G.; Papapetropoulos, A. The Insertion/Deletion Polymorphism of the Angiotensin Converting Enzyme (ACE) in Parkinson’s Disease. Open Neurol. J. 2008, 2, 66–70. [Google Scholar] [CrossRef]
- Izzicupo, P.; Ghinassi, B.; D’Amico, M.A.; Di Blasio, A.; Gesi, M.; Napolitano, G.; Gallina, S.; Di Baldassarre, A. Effects of ACE I/D Polymorphism and Aerobic Training on the Immune–Endocrine Network and Cardiovascular Parameters of Postmenopausal Women. J. Clin. Endocrinol. Metab. 2013, 98, 4187–4194. [Google Scholar] [CrossRef]
- Gaggi, G.; Di Credico, A.; Izzicupo, P.; Iannetti, G.; Di Baldassarre, A.; Ghinassi, B. Chemical and Biological Molecules Involved in Differentiation, Maturation, and Survival of Dopaminergic Neurons in Health and Parkinson’s Disease: Physiological Aspects and Clinical Implications. Biomedicines 2021, 9, 754. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein Misfolding and Aggregation: Implications in Parkinson’s Disease Pathogenesis. Biochim. Biophys. Acta BBA—Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Kanazawa, T.; Adachi, E.; Orimo, S.; Nakamura, A.; Mizusawa, H.; Uchihara, T. Pale Neurites, Premature α-Synuclein Aggregates with Centripetal Extension from Axon Collaterals. Brain Pathol. 2012, 22, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xie, X.; Liu, R. The Role of α-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef] [PubMed]
- Moreno-García, A.; Kun, A.; Calero, M.; Calero, O. The Neuromelanin Paradox and Its Dual Role in Oxidative Stress and Neurodegeneration. Antioxidants 2021, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of Iron, Dopamine and Neuromelanin Pathways in Brain Aging and Parkinson’s Disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-L.; Chen, Y.; Zhang, C.-H.; Wang, Y.-X.; Fernandez-Funez, P. Genetics of Parkinson’s Disease and Related Disorders. J. Med. Genet. 2018, 55, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Ai, S.; Xu, Q.; Hu, Y.; Song, C.; Guo, J.; Shen, L.; Wang, C.; Yu, R.; Yan, X.; Tang, B. Hypomethylation of SNCA in Blood of Patients with Sporadic Parkinson’s Disease. J. Neurol. Sci. 2014, 337, 123–128. [Google Scholar] [CrossRef]
- Pavlou, M.A.S.; Pinho, R.; Paiva, I.; Outeiro, T.F. The Yin and Yang of α-Synuclein-Associated Epigenetics in Parkinson’s Disease. Brain 2016, 140, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Júnior, H.J.; Bucci, C.; Marzetti, E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules 2021, 11, 1508. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. α-Synuclein and Mechanisms of Epigenetic Regulation. Brain Sci. 2023, 13, 150. [Google Scholar] [CrossRef] [PubMed]
- Pieper, H.C.; Evert, B.O.; Kaut, O.; Riederer, P.F.; Waha, A.; Wüllner, U. Different Methylation of the TNF-Alpha Promoter in Cortex and Substantia Nigra: Implications for Selective Neuronal Vulnerability. Neurobiol. Dis. 2008, 32, 521–527. [Google Scholar] [CrossRef]
- Rasheed, M.; Liang, J.; Wang, C.; Deng, Y.; Chen, Z. Epigenetic Regulation of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4956. [Google Scholar] [CrossRef]
- Wen, K.; Miliç, J.; El-Khodor, B.; Dhana, K.; Nano, J.; Pulido, T.; Kraja, B.; Zaciragic, A.; Bramer, W.M.; Troup, J.; et al. The Role of DNA Methylation and Histone Modifications in Neurodegenerative Diseases: A Systematic Review. PLoS ONE 2016, 11, e0167201. [Google Scholar] [CrossRef] [PubMed]
- Rubino, A.; D’Addario, C.; Di Bartolomeo, M.; Michele Salamone, E.; Locuratolo, N.; Fattapposta, F.; Vanacore, N.; Pascale, E. DNA Methylation of the 5′-UTR DAT 1 Gene in Parkinson’s Disease Patients. Acta Neurol. Scand. 2020, 142, 275–280. [Google Scholar] [CrossRef]
- Kochmanski, J.; Kuhn, N.C.; Bernstein, A.I. Parkinson’s Disease-Associated, Sex-Specific Changes in DNA Methylation at PARK7 (DJ-1), SLC17A6 (VGLUT2), PTPRN2 (IA-2β), and NR4A2 (NURR1) in Cortical Neurons. Npj Park. Dis. 2022, 8, 120. [Google Scholar] [CrossRef]
- Min, S.; Xu, Q.; Qin, L.; Li, Y.; Li, Z.; Chen, C.; Wu, H.; Han, J.; Zhu, X.; Jin, P.; et al. Altered Hydroxymethylome in the Substantia Nigra of Parkinson’s Disease. Hum. Mol. Genet. 2022, 31, 3494–3503. [Google Scholar] [CrossRef]
- Renani, P.G.; Taheri, F.; Rostami, D.; Farahani, N.; Abdolkarimi, H.; Abdollahi, E.; Taghizadeh, E.; Gheibi Hayat, S.M. Involvement of Aberrant Regulation of Epigenetic Mechanisms in the Pathogenesis of Parkinson’s Disease and Epigenetic-based Therapies. J. Cell. Physiol. 2019, 234, 19307–19319. [Google Scholar] [CrossRef]
- Toker, L.; Tran, G.T.; Sundaresan, J.; Tysnes, O.-B.; Alves, G.; Haugarvoll, K.; Nido, G.S.; Dölle, C.; Tzoulis, C. Genome-Wide Histone Acetylation Analysis Reveals Altered Transcriptional Regulation in the Parkinson’s Disease Brain. Mol. Neurodegener. 2021, 16, 31. [Google Scholar] [CrossRef]
- Gebremedhin, K.G.; Rademacher, D.J. Histone H3 Acetylation in the Postmortem Parkinson’s Disease Primary Motor Cortex. Neurosci. Lett. 2016, 627, 121–125. [Google Scholar] [CrossRef]
- Lu, C.; Ren, S.; Xie, W.; Zhao, Z.; Wu, X.; Guo, S.; Suo, A.; Zhou, N.; Yang, J.; Wu, S.; et al. Characterizing Relevant MicroRNA Editing Sites in Parkinson’s Disease. Cells 2022, 12, 75. [Google Scholar] [CrossRef]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA Feedback Circuit in Midbrain Dopamine Neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef]
- Li, J.; Dani, J.A.; Le, W. The Role of Transcription Factor Pitx3 in Dopamine Neuron Development and Parkinson’s Disease. Curr. Top. Med. Chem. 2009, 9, 855–859. [Google Scholar] [PubMed]
- Zhang, H.-Q.; Wang, J.-Y.; Li, Z.-F.; Cui, L.; Huang, S.-S.; Zhu, L.-B.; Sun, Y.; Yang, R.; Fan, H.-H.; Zhang, X.; et al. DNA Methyltransferase 1 Is Dysregulated in Parkinson’s Disease via Mediation of miR-17. Mol. Neurobiol. 2021, 58, 2620–2633. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Urso, D.; Tortelli, R. The Challenge of Amyotrophic Lateral Sclerosis Descriptive Epidemiology: To Estimate Low Incidence Rates across Complex Phenotypes in Different Geographic Areas. Curr. Opin. Neurol. 2022, 35, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primer 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic Lateral Sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Rosenbohm, A.; Schmid, B.; Buckert, D.; Rottbauer, W.; Kassubek, J.; Ludolph, A.C.; Bernhardt, P. Cardiac Findings in Amyotrophic Lateral Sclerosis: A Magnetic Resonance Imaging Study. Front. Neurol. 2017, 8, 479. [Google Scholar] [CrossRef] [PubMed]
- Gaggi, G.; Di Credico, A.; Izzicupo, P.; Sancilio, S.; Di Mauro, M.; Iannetti, G.; Dolci, S.; Amabile, G.; Di Baldassarre, A.; Ghinassi, B. Decellularized Extracellular Matrices and Cardiac Differentiation: Study on Human Amniotic Fluid-Stem Cells. Int. J. Mol. Sci. 2020, 21, 6317. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Pacheco, A.; Franco, J.M.; Lopez, S.; Gomez-Zumaquero, J.M.; Magdalena Leal-Lasarte, M.; Caballero-Hernandez, D.E.; Cejudo-Guillén, M.; Pozo, D. Epigenetic Mechanisms of Gene Regulation in Amyotrophic Lateral Sclerosis. In Neuroepigenomics in Aging and Disease; Delgado-Morales, R., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 978, pp. 255–275. ISBN 978-3-319-53888-4. [Google Scholar]
- Caballero-Hernandez, D.; Toscano, M.G.; Cejudo-Guillen, M.; Garcia-Martin, M.L.; Lopez, S.; Franco, J.M.; Quintana, F.J.; Roodveldt, C.; Pozo, D. The ‘Omics’ of Amyotrophic Lateral Sclerosis. Trends Mol. Med. 2016, 22, 53–67. [Google Scholar] [CrossRef]
- PARALS Registry; SLALOM Group; SLAP Registry; FALS Sequencing Consortium; SLAGEN Consortium; NNIPPS Study Group; Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; et al. Genome-Wide Association Analyses Identify New Risk Variants and the Genetic Architecture of Amyotrophic Lateral Sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells 2020, 9, 2413. [Google Scholar] [CrossRef]
- Cozzi, M.; Ferrari, V. Autophagy Dysfunction in ALS: From Transport to Protein Degradation. J. Mol. Neurosci. 2022, 72, 1456–1481. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.-G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef] [PubMed]
- Lépine, S.; Castellanos-Montiel, M.J.; Durcan, T.M. TDP-43 Dysregulation and Neuromuscular Junction Disruption in Amyotrophic Lateral Sclerosis. Transl. Neurodegener. 2022, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.; Duddy, W.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.J.; Park, N.Y.; Dubey, S.K.; Kim, T.; Ruan, K.; Lim, S.B.; Park, S.-H.; Ha, S.; Kovlyagina, I.; et al. Multi-Omic Analysis of Selectively Vulnerable Motor Neuron Subtypes Implicates Altered Lipid Metabolism in ALS. Nat. Neurosci. 2021, 24, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Castanedo-Vazquez, D.; Bosque-Varela, P.; Sainz-Pelayo, A.; Riancho, J. Infectious Agents and Amyotrophic Lateral Sclerosis: Another Piece of the Puzzle of Motor Neuron Degeneration. J. Neurol. 2019, 266, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Cucovici, A.; Fontana, A.; Ivashynka, A.; Russo, S.; Renna, V.; Mazzini, L.; Gagliardi, I.; Mandrioli, J.; Martinelli, I.; Lisnic, V.; et al. The Impact of Lifetime Alcohol and Cigarette Smoking Loads on Amyotrophic Lateral Sclerosis Progression: A Cross-Sectional Study. Life 2021, 11, 352. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Gralinski, L.; Armour, C.D.; Ferris, M.T.; Thomas, M.J.; Proll, S.; Bradel-Tretheway, B.G.; Korth, M.J.; Castle, J.C.; Biery, M.C.; et al. Unique Signatures of Long Noncoding RNA Expression in Response to Virus Infection and Altered Innate Immune Signaling. mBio 2010, 1, e00206-10. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of Epigenetically Altered Genes in Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef]
- Hop, P.J.; Zwamborn, R.A.J.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; Van Rheenen, W.; Van Vugt, J.J.F.A.; Dekker, A.M.; Westeneng, H.-J.; et al. Genome-Wide Study of DNA Methylation Shows Alterations in Metabolic, Inflammatory, and Cholesterol Pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in Amyotrophic Lateral Sclerosis: A Role for Histone Post-Translational Modifications in Neurodegenerative Disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Ortega, J.A.; Quinlan, K.A.; Santos, D.P.; Gu, H.; Martin, E.J.; Galonska, C.; Pop, R.; Maidl, S.; Di Pardo, A.; et al. Dissecting the Functional Consequences of De Novo DNA Methylation Dynamics in Human Motor Neuron Differentiation and Physiology. Cell Stem Cell 2018, 22, 559–574.e9. [Google Scholar] [CrossRef] [PubMed]
- Ozyurt, T.; Gautam, M. Differential Epigenetic Signature of Corticospinal Motor Neurons in ALS. Brain Sci. 2021, 11, 754. [Google Scholar] [CrossRef]
- Klingl, Y.E.; Pakravan, D.; Van Den Bosch, L. Opportunities for Histone Deacetylase Inhibition in Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 2021, 178, 1353–1372. [Google Scholar] [CrossRef] [PubMed]
- Waddell, J.; Banerjee, A.; Kristian, T. Acetylation in Mitochondria Dynamics and Neurodegeneration. Cells 2021, 10, 3031. [Google Scholar] [CrossRef]
- Iaconelli, J.; Xuan, L.; Karmacharya, R. HDAC6 Modulates Signaling Pathways Relevant to Synaptic Biology and Neuronal Differentiation in Human Stem-Cell-Derived Neurons. Int. J. Mol. Sci. 2019, 20, 1605. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in Human Post-Mortem Amyotrophic Lateral Sclerosis Spinal Cords Provides Insight into Disease Mechanisms. Mol. Cell. Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hawley, Z.C.E.; Campos-Melo, D.; Strong, M.J. Evidence of A Negative Feedback Network Between TDP-43 and miRNAs Dependent on TDP-43 Nuclear Localization. J. Mol. Biol. 2020, 432, 166695. [Google Scholar] [CrossRef]
- Di Pietro, L.; Lattanzi, W.; Bernardini, C. Skeletal Muscle MicroRNAs as Key Players in the Pathogenesis of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2018, 19, 1534. [Google Scholar] [CrossRef]
- Koike, Y.; Onodera, O. Implications of miRNAs Dysregulation in Amyotrophic Lateral Sclerosis: Challenging for Clinical Applications. Front. Neurosci. 2023, 17, 1131758. [Google Scholar] [CrossRef] [PubMed]
- Nie, M.; Deng, Z.-L.; Liu, J.; Wang, D.-Z. Noncoding RNAs, Emerging Regulators of Skeletal Muscle Development and Diseases. BioMed Res. Int. 2015, 2015, 676575. [Google Scholar] [CrossRef] [PubMed]
- Jamebozorgi, K.; Rostami, D.; Pormasoumi, H.; Taghizadeh, E.; Barreto, G.E.; Sahebkar, A. Epigenetic Aspects of Multiple Sclerosis and Future Therapeutic Options. Int. J. Neurosci. 2021, 131, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple Sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Doshi, A.; Chataway, J. Multiple Sclerosis, a Treatable Disease. Clin. Med. 2017, 17, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Hachim, M.; Elemam, N.; Maghazachi, A. The Beneficial and Debilitating Effects of Environmental and Microbial Toxins, Drugs, Organic Solvents and Heavy Metals on the Onset and Progression of Multiple Sclerosis. Toxins 2019, 11, 147. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.S.-F. Epigenetics in Multiple Sclerosis. In Epigenetics in Allergy and Autoimmunity; Chang, C., Lu, Q., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2020; Volume 1253, pp. 309–374. ISBN 9789811534485. [Google Scholar]
- Lossius, A.; Johansen, J.; Torkildsen, Ø.; Vartdal, F.; Holmøy, T. Epstein-Barr Virus in Systemic Lupus Erythematosus, Rheumatoid Arthritis and Multiple Sclerosis—Association and Causation. Viruses 2012, 4, 3701–3730. [Google Scholar] [CrossRef] [PubMed]
- Riise, T.; Nortvedt, M.W.; Ascherio, A. Smoking Is a Risk Factor for Multiple Sclerosis. Neurology 2003, 61, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Scazzone, C.; Agnello, L.; Bivona, G.; Lo Sasso, B.; Ciaccio, M. Vitamin D and Genetic Susceptibility to Multiple Sclerosis. Biochem. Genet. 2021, 59, 1–30. [Google Scholar] [CrossRef]
- Gacias, M.; Casaccia, P. Epigenetic Mechanisms in Multiple Sclerosis. Rev. Esp. Escler. Mult. 2014, 6, 25–35. [Google Scholar]
- Tang, Y.; Luo, M.; Pan, K.; Ahmad, T.; Zhou, T.; Miao, Z.; Zhou, H.; Sun, H.; Xu, X.; Namaka, M.; et al. DNA Hydroxymethylation Changes in Response to Spinal Cord Damage in a Multiple Sclerosis Mouse Model. Epigenomics 2019, 11, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.W.; Metz, L.M.; Kovalchuk, O. Epigenetic Changes in Patients with Multiple Sclerosis. Nat. Rev. Neurol. 2013, 9, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyakova, G.Y.; Piket, E.; Marabita, F.; Pahlevan Kakhki, M.; Ewing, E.; Ruhrmann, S.; Needhamsen, M.; Jagodic, M.; Kular, L. Epigenetic Research in Multiple Sclerosis: Progress, Challenges, and Opportunities. Physiol. Genom. 2017, 49, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Tegla, C.A.; Azimzadeh, P.; Andrian-Albescu, M.; Martin, A.; Cudrici, C.D.; Trippe, R.; Sugarman, A.; Chen, H.; Boodhoo, D.; Vlaicu, S.I.; et al. SIRT1 Is Decreased during Relapses in Patients with Multiple Sclerosis. Exp. Mol. Pathol. 2014, 96, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kular, L.; Jagodic, M. Epigenetic Insights into Multiple Sclerosis Disease Progression. J. Intern. Med. 2020, 288, 82–102. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.K.; Li, S.; Arning, E.; Alkhayer, K.; Clements, R.; Sarcyk, Z.; Dassanayake, R.S.; Brasch, N.E.; Freeman, E.J.; Bottiglieri, T.; et al. Changes in Methionine Metabolism and Histone H3 Trimethylation Are Linked to Mitochondrial Defects in Multiple Sclerosis. J. Neurosci. 2015, 35, 15170–15186. [Google Scholar] [CrossRef] [PubMed]
- Junker, A.; Hohlfeld, R.; Meinl, E. The Emerging Role of microRNAs in Multiple Sclerosis. Nat. Rev. Neurol. 2011, 7, 56–59. [Google Scholar] [CrossRef]
- Du, C.; Liu, C.; Kang, J.; Zhao, G.; Ye, Z.; Huang, S.; Li, Z.; Wu, Z.; Pei, G. MicroRNA miR-326 Regulates TH-17 Differentiation and Is Associated with the Pathogenesis of Multiple Sclerosis. Nat. Immunol. 2009, 10, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, F.; Ellestad, K.K.; Maingat, F.; Warren, K.G.; Han, M.H.; Steinman, L.; Baker, G.B.; Power, C. Impaired Neurosteroid Synthesis in Multiple Sclerosis. Brain 2011, 134, 2703–2721. [Google Scholar] [CrossRef]
- Gaggi, G.; Izzicupo, P.; Di Credico, A.; Sancilio, S.; Di Baldassarre, A.; Ghinassi, B. Spare Parts from Discarded Materials: Fetal Annexes in Regenerative Medicine. Int. J. Mol. Sci. 2019, 20, 1573. [Google Scholar] [CrossRef]
- Barbaccia, M.L. Neurosteroidogenesis: Relevance to Neurosteroid Actions in Brain and Modulation by Psychotropic Drugs. Crit. Rev. Neurobiol. 2004, 16, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 Cluster: A Comprehensive Update on Its Genomics, Genetics, Functions and Increasingly Important and Numerous Roles in Health and Disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef]
- Liu, S.-Q.; Jiang, S.; Li, C.; Zhang, B.; Li, Q.-J. miR-17-92 Cluster Targets Phosphatase and Tensin Homology and Ikaros Family Zinc Finger 4 to Promote TH17-Mediated Inflammation. J. Biol. Chem. 2014, 289, 12446–12456. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.B.; Cairns, M.J.; Gandhi, K.S.; Carroll, A.P.; Moscovis, S.; Stewart, G.J.; Broadley, S.; Scott, R.J.; Booth, D.R.; Lechner-Scott, J.; et al. MicroRNAs miR-17 and miR-20a Inhibit T Cell Activation Genes and Are Under-Expressed in MS Whole Blood. PLoS ONE 2010, 5, e12132. [Google Scholar] [CrossRef]
- Lindberg, R.L.P.; Hoffmann, F.; Mehling, M.; Kuhle, J.; Kappos, L. Altered Expression of miR-17-5p in CD4+ Lymphocytes of Relapsing–Remitting Multiple Sclerosis Patients. Eur. J. Immunol. 2010, 40, 888–898. [Google Scholar] [CrossRef]
- Shusharina, N.; Yukhnenko, D.; Botman, S.; Sapunov, V.; Savinov, V.; Kamyshov, G.; Sayapin, D.; Voznyuk, I. Modern Methods of Diagnostics and Treatment of Neurodegenerative Diseases and Depression. Diagnostics 2023, 13, 573. [Google Scholar] [CrossRef]
- Coppede, F. Targeting the Epigenome to Treat Neurodegenerative Diseases or Delay Their Onset: A Perspective. Neural Regen. Res. 2022, 17, 1745. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in Human Disease and Prospects for Epigenetic Therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xie, M.; Xue, J.; Xiang, L.; Li, Y.; Xiao, J.; Xiao, G.; Wang, H. EGCG Ameliorates Neuronal and Behavioral Defects by Remodeling Gut Microbiota and TotM Expression in Drosophila Models of Parkinson’s Disease. FASEB J. 2020, 34, 5931–5950. [Google Scholar] [CrossRef]
- Teng, Y.; Zhao, J.; Ding, L.; Ding, Y.; Zhou, P. Complex of EGCG with Cu(II) Suppresses Amyloid Aggregation and Cu(II)-Induced Cytotoxicity of α-Synuclein. Molecules 2019, 24, 2940. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Wang, I.-H.; Rajesh, R. Use of Leptin-Conjugated Phosphatidic Acid Liposomes with Resveratrol and Epigallocatechin Gallate to Protect Dopaminergic Neurons against Apoptosis for Parkinson’s Disease Therapy. Acta Biomater. 2021, 119, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, J.; Wang, Y.; Wang, G.; Tang, P.; Liu, Y.; Zhang, Y.; Ouyang, L. Targeting Epigenetic Modifications in Parkinson’s Disease Therapy. Med. Res. Rev. 2023, 43, 1748–1777. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, P.S.; Dallas, S.; Wilson, B.; Block, M.L.; Wang, C.-C.; Kinyamu, H.; Lu, N.; Gao, X.; Leng, Y.; et al. Histone Deacetylase Inhibitors Up-Regulate Astrocyte GDNF and BDNF Gene Transcription and Protect Dopaminergic Neurons. Int. J. Neuropsychopharmacol. 2008, 11, 1123. [Google Scholar] [CrossRef]
- Teijido, O.; Cacabelos, R. Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2018, 19, 3199. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, M.; Goulding, S.R.; Wyatt, S.L.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. LMK235, a Small Molecule Inhibitor of HDAC4/5, Protects Dopaminergic Neurons against Neurotoxin- and α-Synuclein-Induced Degeneration in Cellular Models of Parkinson’s Disease. Mol. Cell. Neurosci. 2021, 115, 103642. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Hsu, P.; Chang, W.; Yu, C.; Wang, Y.; Yang, J.; Tsai, F.; Chen, K.; Tsai, C.; Bau, D. Protective Effects of Valproic Acid on 6-hydroxydopamine-induced Neuroinjury. Environ. Toxicol. 2020, 35, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, M.; Du, R.-H.; Qiao, C.; Jiang, C.-Y.; Zhang, K.-Z.; Ding, J.-H.; Hu, G. MicroRNA-7 Targets Nod-like Receptor Protein 3 Inflammasome to Modulate Neuroinflammation in the Pathogenesis of Parkinson’s Disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-B.; Zhang, Y.-F.; Wang, H.; Ren, R.-J.; Cui, H.-L.; Huang, W.-Y.; Cheng, Q.; Chen, H.-Z.; Wang, G. miR-425 Deficiency Promotes Necroptosis and Dopaminergic Neurodegeneration in Parkinson’s Disease. Cell Death Dis. 2019, 10, 589. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Lin, T.-K.; Huang, Y.-H. MiR-29a Inhibits MPP +—Induced Cell Death and Inflammation in Parkinson’s Disease Model in Vitro by Potential Targeting of MAVS. Eur. J. Pharmacol. 2022, 934, 175302. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; DeWitt, M.R.; Bornhorst, J.; Soares, F.A.; Mukhopadhyay, S.; Bowman, A.B.; Aschner, M. Age- and Manganese-Dependent Modulation of Dopaminergic Phenotypes in a C. elegans DJ-1 Genetic Model of Parkinson’s Disease. Metallomics 2015, 7, 289–298. [Google Scholar] [CrossRef]
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-α Regulates miRNA Targeting Mitochondrial Complex-I and Induces Cell Death in Dopaminergic Cells. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2015, 1852, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Xiong, J.; Ji, L.; Xue, X. MiR-421 Aggravates Neurotoxicity and Promotes Cell Death in Parkinson’s Disease Models by Directly Targeting MEF2D. Neurochem. Res. 2021, 46, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Brown, R.H.; et al. Sodium Phenylbutyrate Prolongs Survival and Regulates Expression of Anti-apoptotic Genes in Transgenic Amyotrophic Lateral Sclerosis Mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Rouaux, C.; Panteleeva, I.; René, F.; Gonzalez De Aguilar, J.-L.; Echaniz-Laguna, A.; Dupuis, L.; Menger, Y.; Boutillier, A.-L.; Loeffler, J.-P. Sodium Valproate Exerts Neuroprotective Effects In Vivo through CREB-Binding Protein-Dependent Mechanisms But Does Not Improve Survival in an Amyotrophic Lateral Sclerosis Mouse Model. J. Neurosci. 2007, 27, 5535–5545. [Google Scholar] [CrossRef] [PubMed]
- Kuta, R.; Larochelle, N.; Fernandez, M.; Pal, A.; Minotti, S.; Tibshirani, M.; St. Louis, K.; Gentil, B.J.; Nalbantoglu, J.N.; Hermann, A.; et al. Depending on the Stress, Histone Deacetylase Inhibitors Act as Heat Shock Protein Co-Inducers in Motor Neurons and Potentiate Arimoclomol, Exerting Neuroprotection through Multiple Mechanisms in ALS Models. Cell Stress Chaperones 2020, 25, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.-E.; Ko, C.-P. Treatment with Trichostatin A Initiated after Disease Onset Delays Disease Progression and Increases Survival in a Mouse Model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Tzeplaeff, L.; Wilfling, S.; Requardt, M.V.; Herdick, M. Current State and Future Directions in the Therapy of ALS. Cells 2023, 12, 1523. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Peedicayil, J. Epigenetic Drugs for Multiple Sclerosis. Curr. Neuropharmacol. 2016, 14, 3–9. [Google Scholar] [CrossRef]
- Mangano, K.; Fagone, P.; Bendtzen, K.; Meroni, P.L.; Quattrocchi, C.; Mammana, S.; Di Rosa, M.; Malaguarnera, L.; Coco, M.; Magro, G.; et al. Hypomethylating Agent 5-Aza-2′-deoxycytidine (DAC) Ameliorates Multiple Sclerosis in Mouse Models. J. Cell. Physiol. 2014, 229, 1918–1925. [Google Scholar] [CrossRef]
- Camelo, S.; Iglesias, A.H.; Hwang, D.; Due, B.; Ryu, H.; Smith, K.; Gray, S.G.; Imitola, J.; Duran, G.; Assaf, B.; et al. Transcriptional Therapy with the Histone Deacetylase Inhibitor Trichostatin A Ameliorates Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2005, 164, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; et al. Vorinostat, a Histone Deacetylase Inhibitor, Suppresses Dendritic Cell Function and Ameliorates Experimental Autoimmune Encephalomyelitis. Exp. Neurol. 2013, 241, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Moyano, A.L.; Ma, Z.; Deng, Y.; Lin, Y.; Zhao, C.; Zhang, L.; Jiang, M.; He, X.; Ma, Z.; et al. miR-219 Cooperates with miR-338 in Myelination and Promotes Myelin Repair in the CNS. Dev. Cell 2017, 40, 566–582.e5. [Google Scholar] [CrossRef] [PubMed]
- Morquette, B.; Juźwik, C.A.; Drake, S.S.; Charabati, M.; Zhang, Y.; Lécuyer, M.-A.; Galloway, D.A.; Dumas, A.; De Faria Junior, O.; Paradis-Isler, N.; et al. MicroRNA-223 Protects Neurons from Degeneration in Experimental Autoimmune Encephalomyelitis. Brain 2019, 142, 2979–2995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Tian, A.; Wang, J.; Shen, X.; Qi, G.; Tang, Y. miR26a Modulates Th17/Treg Balance in the EAE Model of Multiple Sclerosis by Targeting IL6. NeuroMolecular Med. 2015, 17, 24–34. [Google Scholar] [CrossRef]
- Dolati, S.; Aghebati-Maleki, L.; Ahmadi, M.; Marofi, F.; Babaloo, Z.; Ayramloo, H.; Jafarisavari, Z.; Oskouei, H.; Afkham, A.; Younesi, V.; et al. Nanocurcumin Restores Aberrant miRNA Expression Profile in Multiple Sclerosis, Randomized, Double-blind, Placebo-controlled Trial. J. Cell. Physiol. 2018, 233, 5222–5230. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Martino, P.; Marcozzi, V.; Bibbò, S.; Ghinassi, B.; Di Baldassarre, A.; Gaggi, G.; Di Credico, A. Unraveling the Epigenetic Landscape: Insights into Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis. Brain Sci. 2024, 14, 553. https://doi.org/10.3390/brainsci14060553
Di Martino P, Marcozzi V, Bibbò S, Ghinassi B, Di Baldassarre A, Gaggi G, Di Credico A. Unraveling the Epigenetic Landscape: Insights into Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis. Brain Sciences. 2024; 14(6):553. https://doi.org/10.3390/brainsci14060553
Chicago/Turabian StyleDi Martino, Pierpaolo, Valentina Marcozzi, Sandra Bibbò, Barbara Ghinassi, Angela Di Baldassarre, Giulia Gaggi, and Andrea Di Credico. 2024. "Unraveling the Epigenetic Landscape: Insights into Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis" Brain Sciences 14, no. 6: 553. https://doi.org/10.3390/brainsci14060553
APA StyleDi Martino, P., Marcozzi, V., Bibbò, S., Ghinassi, B., Di Baldassarre, A., Gaggi, G., & Di Credico, A. (2024). Unraveling the Epigenetic Landscape: Insights into Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis. Brain Sciences, 14(6), 553. https://doi.org/10.3390/brainsci14060553