Neuroadaptation in Nicotine Addiction: Update on the Sensitization-Homeostasis Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Major Principles of the Sensitization-Homeostasis Model










3. A Review of Recent Literature in Relation to the Sensitization-Homeostasis Model
3.1. The SH Model Holds that the Neural Processes that Produce Addiction Are Initiated Soon after the Onset of Nondaily Smoking, Stipulating that Addiction Is Possible in the Absence of Sustained Moderate Daily Smoking
3.2. The SH Model Stipulated that Neuroadaptations Develop Quickly with Intermittent Nicotine Exposures
3.3. Nicotine Withdrawal Is Present in Nondaily Smokers. This Contradicted the Prevailing Paradigm that Smokers Do Not Experience Withdrawal or Addiction Until They Are Smoking at Least 5 Cigarettes per Day [39,40]
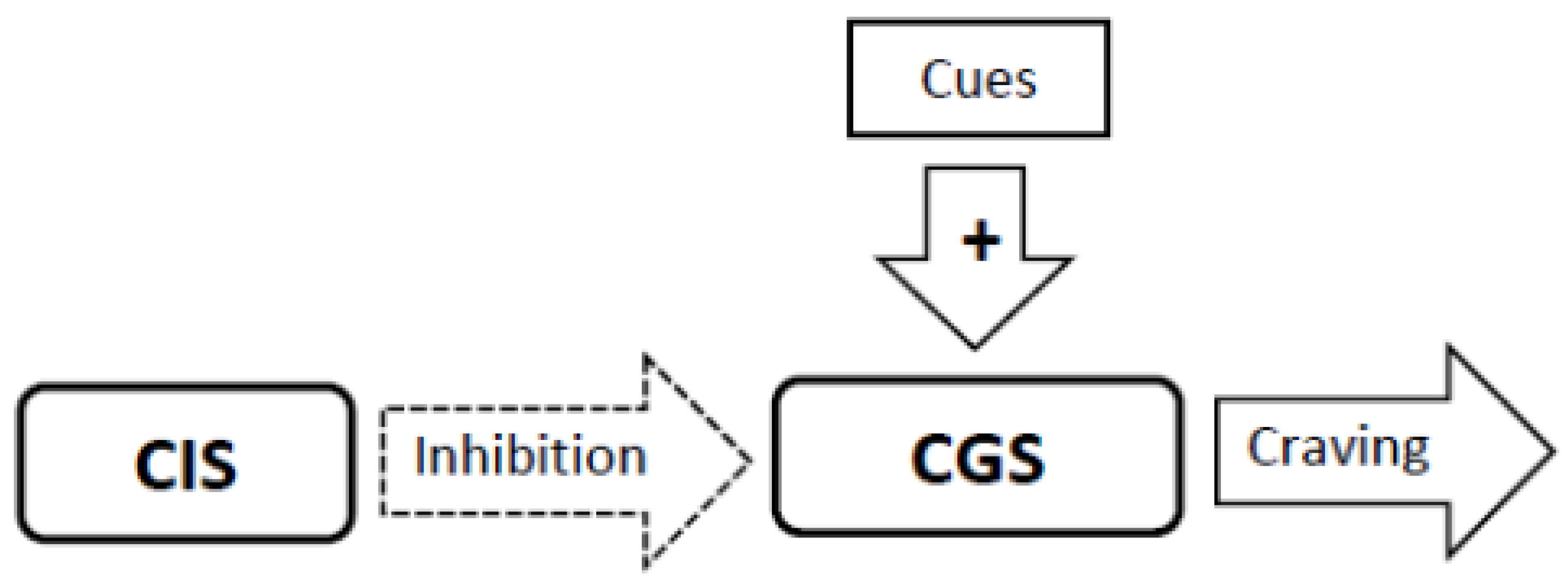
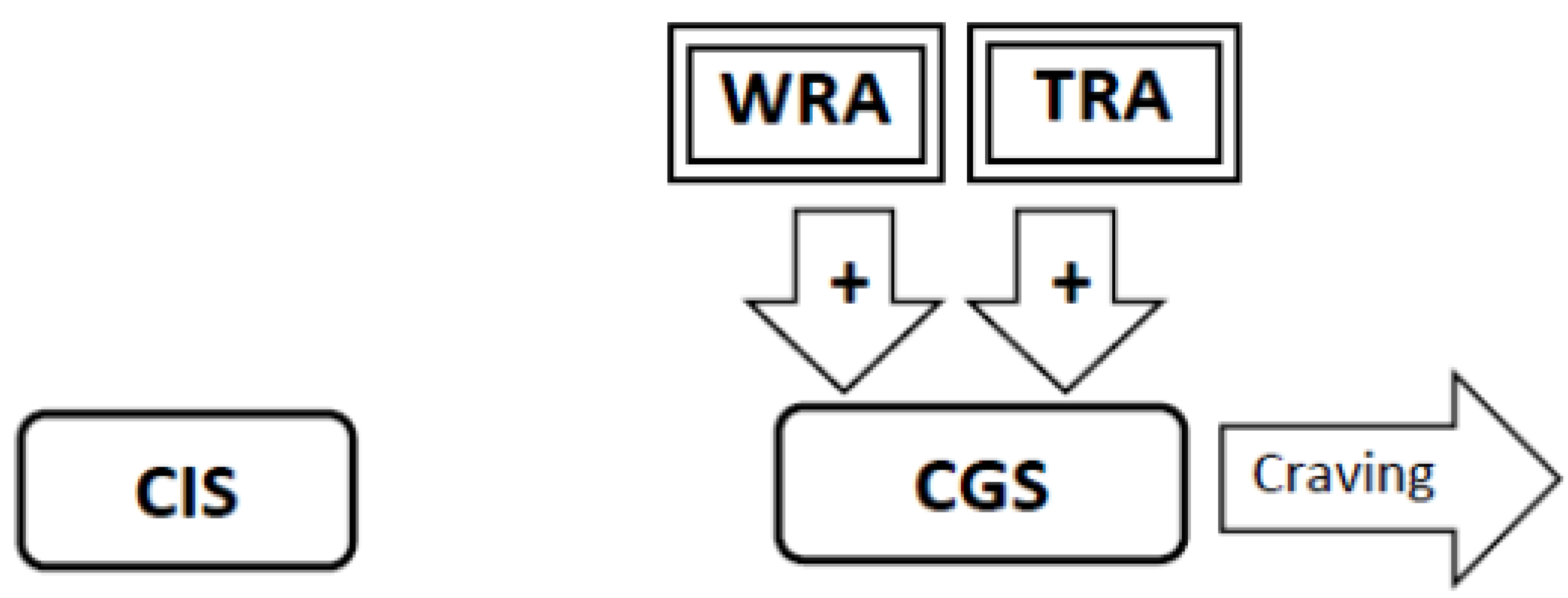
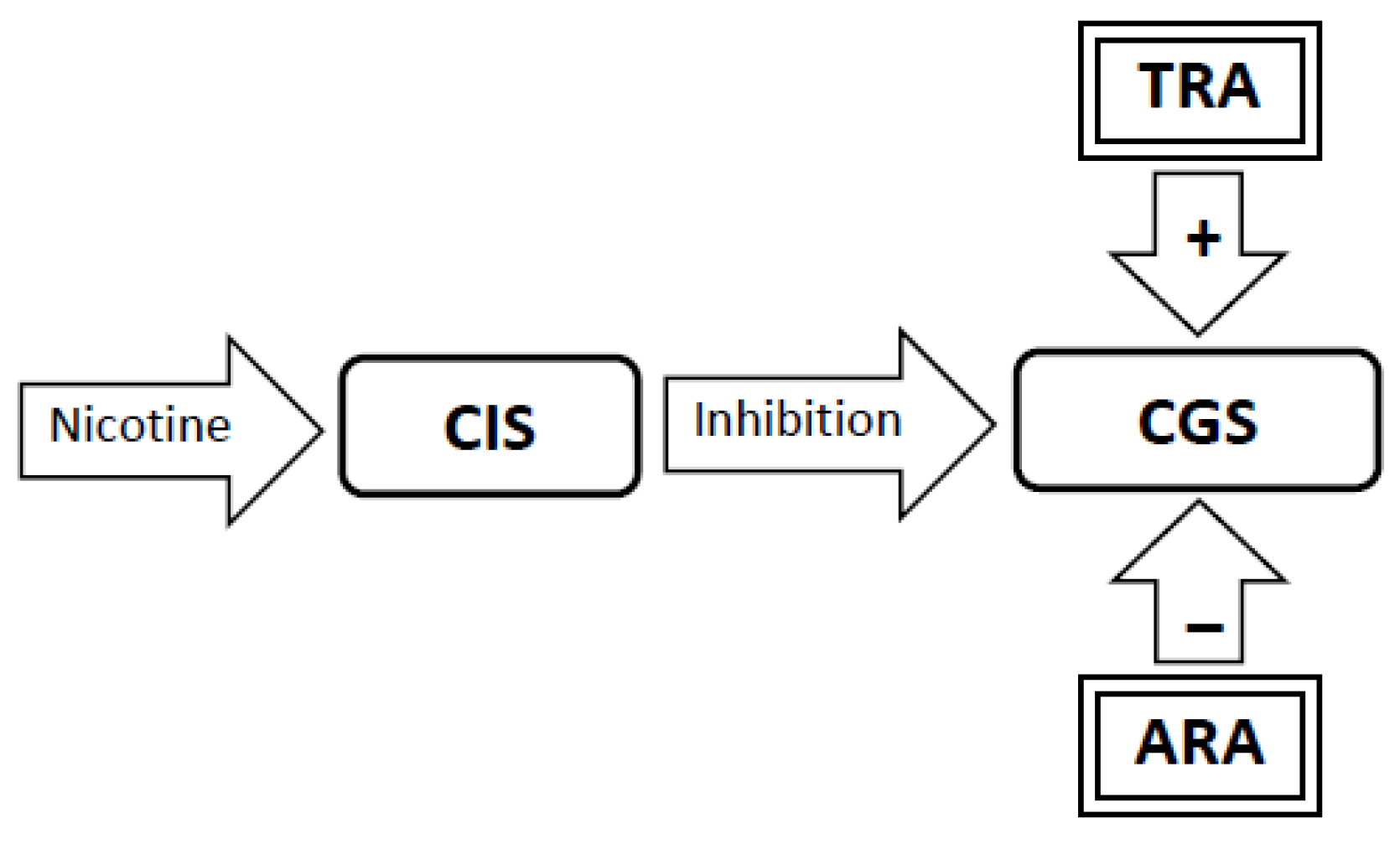
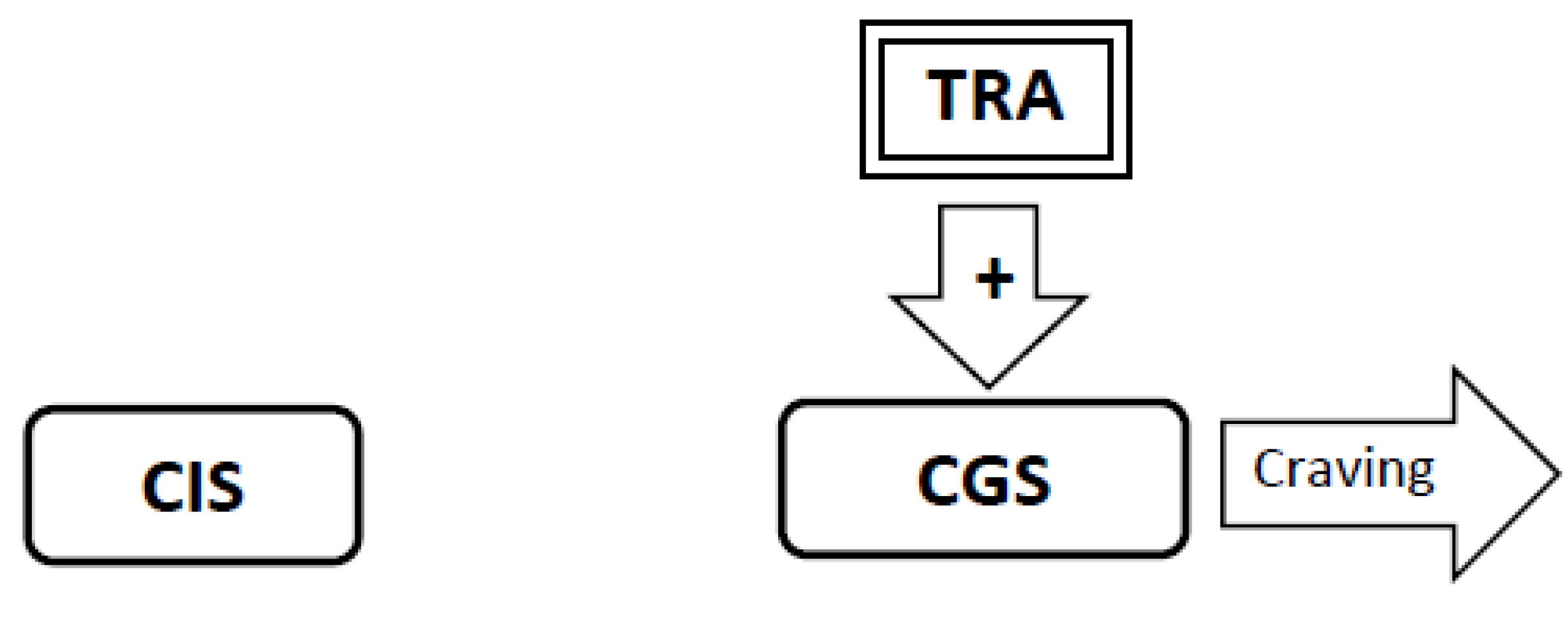
3.4. Experimental Conditions that Stimulate Craving Will Increase Neural Activity in the Craving Generation System (Figure 1)
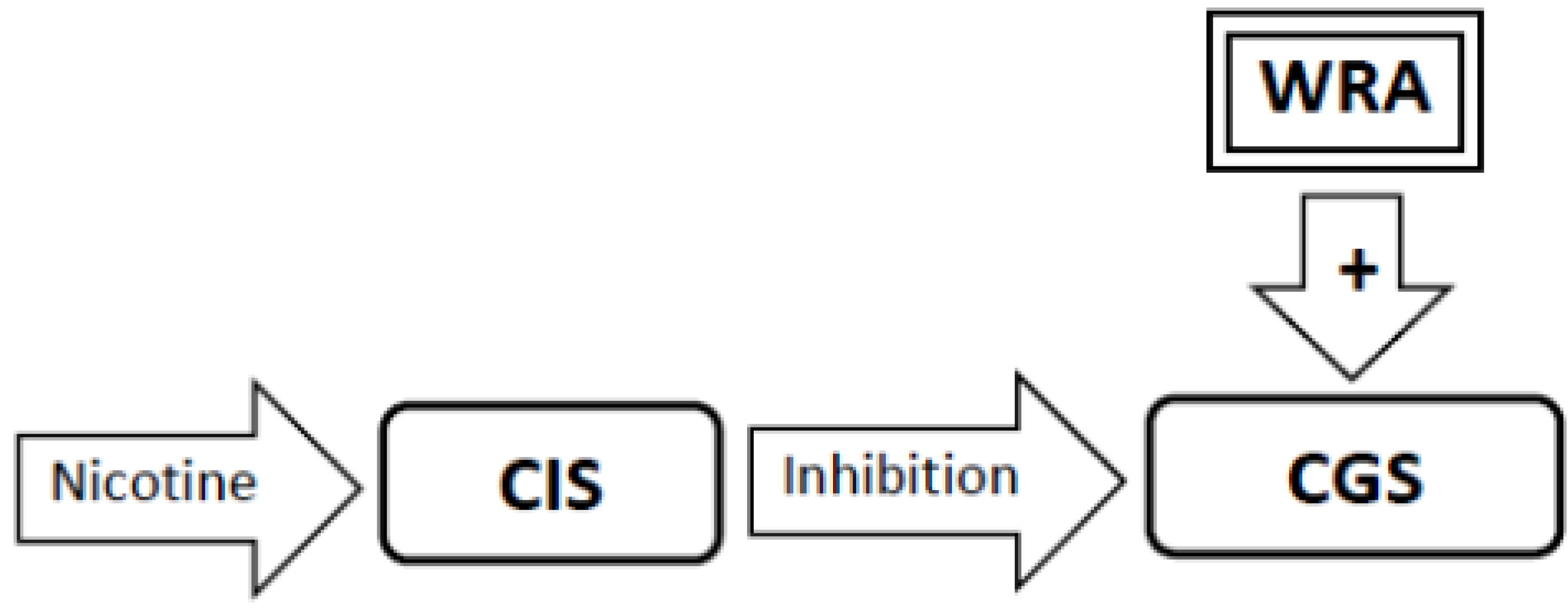
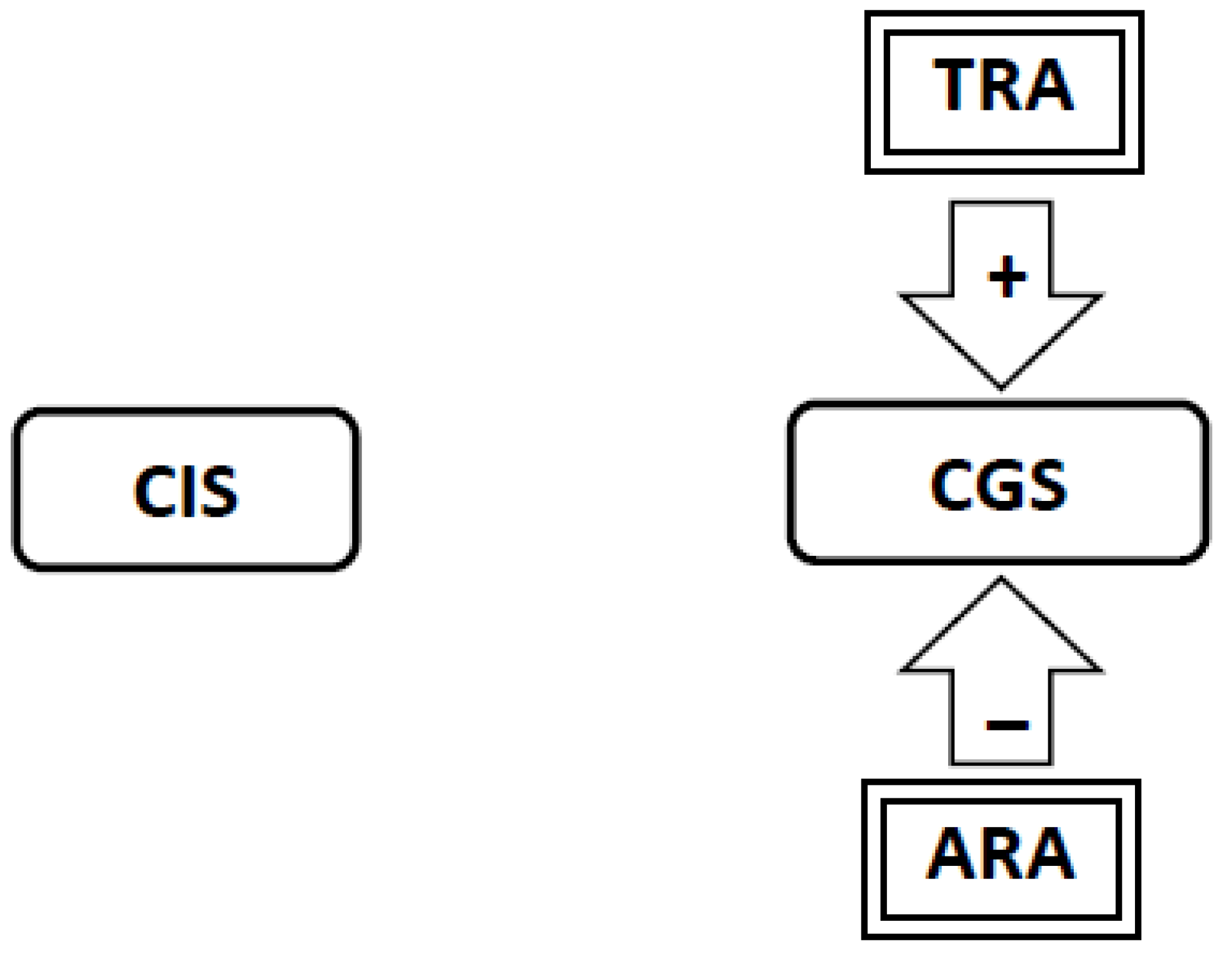
3.5. Nicotine Administration Will Inhibit Activity in the Craving Generation System (Figure 2)
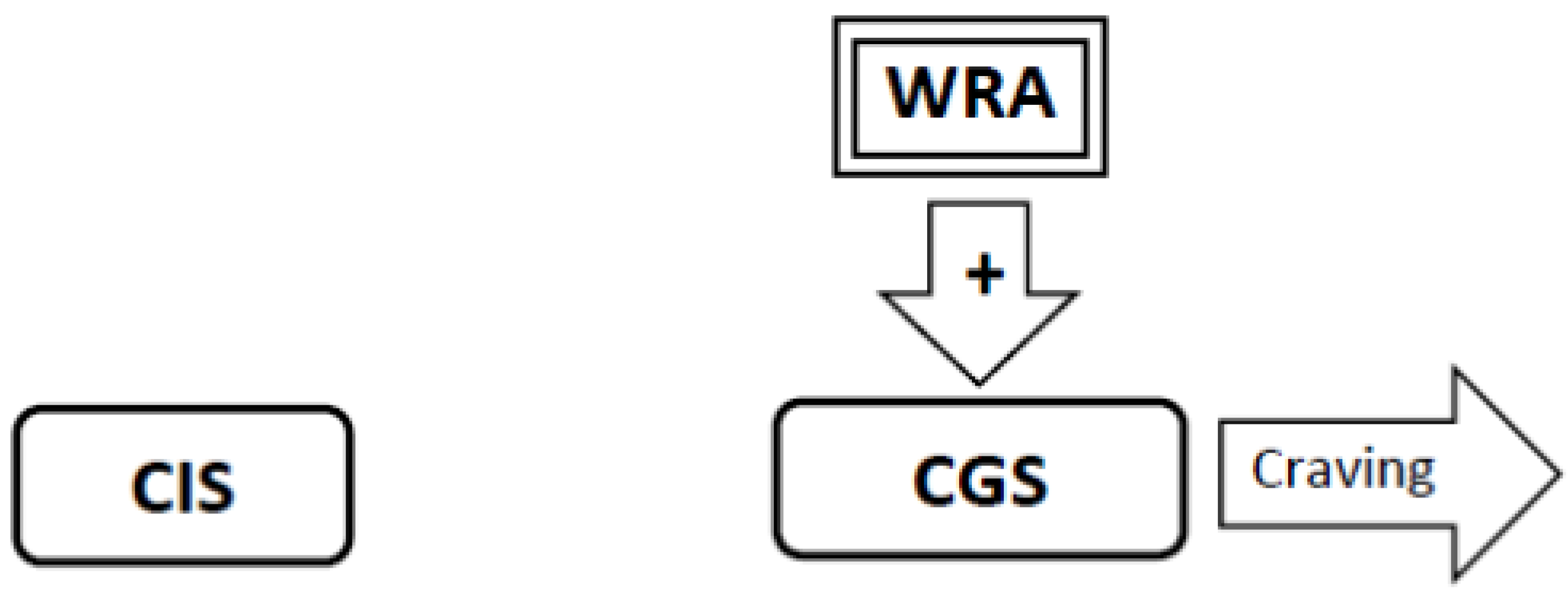
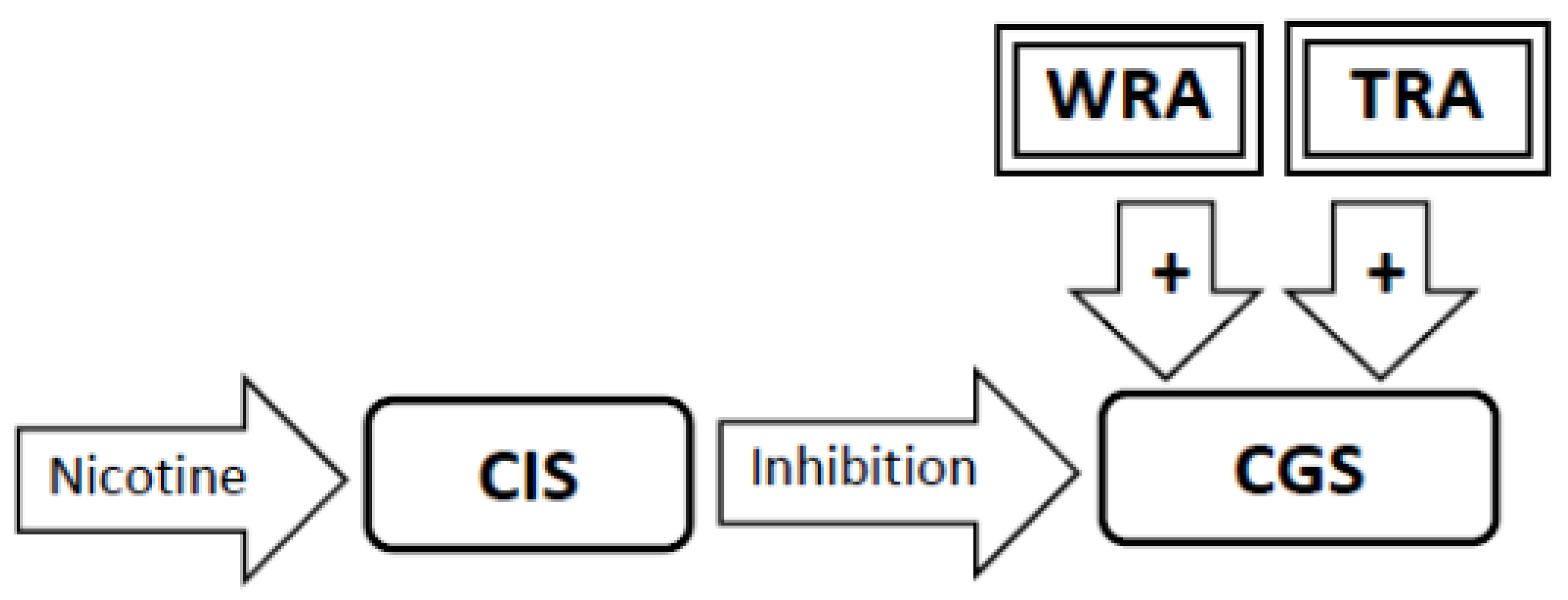
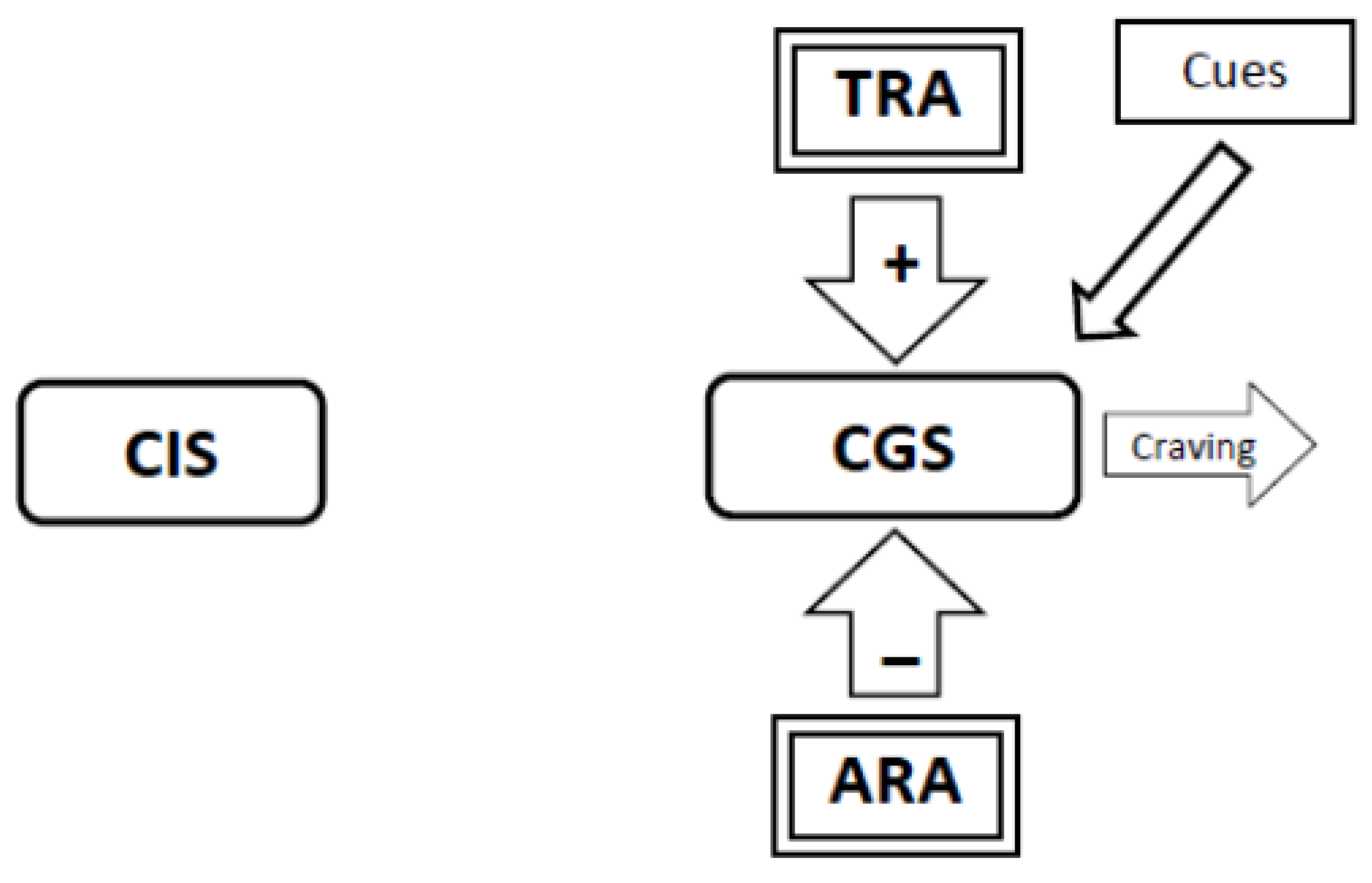
3.6. During Abstinence the Withdrawal-Related Adaptations and Tolerance-Related Adaptations Will Activate the Craving Generation System Resulting in Craving (Figure 4, Figure 5, Figure 6)
3.8. If Tolerance-Related Adaptations Are Irreversible, Successful Smoking Cessation Will Require the Development of Abstinence-Related Adaptations Which Suppress Craving. Continued Abstinence Rests on a Dynamic Equilibrium between Tolerance-Related Adaptations that Stimulate Craving and Opposing Abstinence-Related Adaptations that Suppress Craving (Figure 7)
3.9. When a Long-Abstinent Smoker Slips and Smokes a Cigarette, the Presence of Nicotine Again Disrupts Homeostasis by Suppressing Craving (Figure 9). As the Abstinence-Related Adaptations Also Suppress Craving, Their Presence Augments the Disruption Caused by Nicotine. To Restore Homeostasis, the Brain Dismantles the Abstinence-Related Adaptations (Figure 10). The Lapsed Smoker Will First Experience an Immediate Suppression of Craving Produced by Nicotine, Followed Later by a Resurgence of Craving as the Dismantling of the Abstinence-Related Adaptations Leaves the Withdrawal-Related Adaptations Unopposed, Restoring the Initial State of Addiction. The Intensity of Withdrawal-Induced Craving Triggered by Smoking a Single Cigarette Might Be Nearly as Severe as that Experienced by the Smoker When They Had Quit Smoking. This Causes a Lapse to Turn into a Relapse
4. Updates to the SH model
4.1. The Latency to Withdrawal
4.2. The Levels of Physical Dependence
4.3. What Happens after Cessation?
5. Errors in the Original SH Model
6. Conclusions and Future Directions
Conflict of Interest
References
- Littleton, J. Receptor regulation as a unitary mechanism for drug tolerance and physical dependence-not quite as simple as it seemed! Addiction 2001, 96, 87–101. [Google Scholar] [CrossRef]
- DiFranza, J.R.; Wellman, R.J. A sensitization-homeostasis model of nicotine craving, withdrawal, and tolerance: Integrating the clinical and basic science literature. Nicotine Tob. Res. 2005, 7, 9–26. [Google Scholar] [CrossRef]
- DiFranza, J.R. Implications of the autonomy theory of nicotine dependence. MedGenMed 2002, 4, 8. [Google Scholar]
- O’Loughlin, J.; Gervais, A.; Dugas, E.; Meshefedjian, G. Milestones in the process of cessation among novice adolescent smokers. Am. J. Public Health 2009, 99, 499–504. [Google Scholar]
- Kandel, D.; Hu, M.-C.; Grieisler, P.; Schaffran, C. On the development of nicotine dependence in adolescence. Drug Alcohol Depend. 2007, 91, 26–39. [Google Scholar] [CrossRef]
- DiFranza, J.R.; Savageau, J.A.; Rigotti, N.A.; Fletcher, K.; Ockene, J.K.; McNeill, A.D.; Coleman, M.; Wood, C. Development of symptoms of tobacco dependence in youths: 30 month follow up data from the DANDY study. Tob. Control 2002, 11, 228–235. [Google Scholar] [CrossRef]
- DiFranza, J.; Savageau, J.; Fletcher, K.; O’Loughlin, J.; Pbert, L.; Ockene, J.; McNeill, A.; Hazelton, J.; Friedman, K.; Dussault, G.; et al. Symptoms of tobacco dependence after brief intermittent use—The Development and Assessment of Nicotine Dependence in Youth-2 Study. Arch. Pediatr. Adolesc. Med. 2007, 161, 704–710. [Google Scholar] [CrossRef]
- Dierker, L.; Mermelstein, R. Early emerging nicotine-dependence symptoms: A signal of propensity for chronic smoking behavior in adolescents. J. Pediatr. 2010, 156, 818–822. [Google Scholar] [CrossRef]
- DiFranza, J.R.; Rigotti, N.A.; McNeill, A.D.; Ockene, J.K.; Savageau, J.A.; St Cyr, D.; Coleman, M. Initial symptoms of nicotine dependence in adolescents. Tob. Control 2000, 9, 313–319. [Google Scholar] [CrossRef]
- Scragg, R.; Wellman, R.J.; Laugesen, M.; DiFranza, J.R. Diminished autonomy over tobacco can appear with the first cigarette. Addict. Behav. 2008, 33, 689–698. [Google Scholar] [CrossRef]
- Savageau, J.; Mowery, P.; DiFranza, J. Symptoms of diminished autonomy over cigarettes with non-daily use. Int. J. Environ. Res. Public Health 2009, 6, 25–35. [Google Scholar]
- Caraballo, R.; Novak, S.; Asman, K. Linking quantity/frequency profiles of cigarette smoking to the presence of nicotine dependence symptoms among adolescent smokers: Findings from the 2004 national youth tobacco survey. Nicotine Tob. Res. 2009, 11, 49–57. [Google Scholar] [CrossRef]
- An, L.; Lein, E.; Bliss, R.; Pallonen, U.; Hennrikus, D.; Farley, D.; Hertel, A.; Perry, C.; Lando, H. Loss of Autonomy over Nicotine Use among College Social Smokers. In Proceedings of 10th Annual Meeting of the Society for Research on Nicotine and Tobacco, Rome, Italy, , September 23–26, 2004; Society for Research on Nicotine and Tobacco: Madison, WI, USA, 2004. [Google Scholar]
- Nichter, M.; Nichter, M.; Thompson, P.J.; Shiffman, S.; Moscicki, A.B. Using qualitative research to inform survey development on nicotine dependence among adolescents. Drug Alcohol Depend. 2002, 68, S41–S56. [Google Scholar] [CrossRef]
- Fernando, W.; Wellman, R.; DiFranza, J. The relationship between level of cigarette consumption and latency to the onset of retrospectively reported withdrawal symptoms. Psychopharmacology (Berl.) 2006, 188, 335–342. [Google Scholar]
- O’Loughlin, J.; DiFranza, J.; Tyndale, R.F.; Meshefedjian, G.; McMillan-Davey, E.; Clarke, P.B.; Hanley, J.; Paradis, G. Nicotine-dependence symptoms are associated with smoking frequency in adolescents. Am. J. Prev. Med. 2003, 25, 219–225. [Google Scholar] [CrossRef]
- Storr, C.L. Characteristics associated with rapid transition to tobacco dependence in youth. Nicotine Tob. Res. 2008, 10, 1099–1104. [Google Scholar] [CrossRef]
- Pergadia, M.L.; Heath, A.C.; Martin, N.G.; Madden, P.A. Genetic analyses of DSM-IV nicotine withdrawal in adult twins. Psychol. Med. 2006, 36, 963–972. [Google Scholar] [CrossRef]
- Govind, A.; Walsh, H.; Green, W. Nicotine-induced upregulation of native neuronal nicotinic receptors is caused by multiple mechanisms. J. Neurosci. 2012, 32, 2227–2238. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Addy, N.; Mineur, Y.; Brunzell, D. It is not “either/or”: Activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog. Neurobiol. 2008, 84, 329–342. [Google Scholar] [CrossRef]
- Brody, A.; Mandelkern, M.; London, E.; Olmstead, R.; Farahi, J.; Scheibal, D.; Jou, J.; Allen, V.; Tiongson, E.; Chefer, S.; et al. Cigarette smoking saturates brain alpha-4,beta-2 nicotinic acetylcholine receptors. Arch. Gen. Psychiatry 2006, 63, 907–915. [Google Scholar] [CrossRef]
- Abreu-Villaca, Y.; Seidler, F.J.; Slotkin, T.A. Impact of adolescent nicotine exposure on adenylyl cyclase-mediated cell signaling: Enzyme induction, neurotransmitter-specific effects, regional selectivities, and the role of withdrawal. Brain Res. 2003, 988, 164–172. [Google Scholar] [CrossRef]
- Abreu-Villaca, Y.A.; Seidler, F.J.; Qiao, D.; Tate, C.A.; Cousins, M.M.; Thillai, I.; Slotkin, T.A. Short-term adolescent nicotine exposure has immediate and persistent effects on cholinergic systems: Critical periods, patterns of exposure, dose thresholds. Neuropsychopharmacology 2003, 28, 1935–1949. [Google Scholar] [CrossRef]
- Placzek, A.N.; Zhang, T.A.; Dani, J.A. Age dependent nicotinic influences over dopamine neuron synaptic plasticity. Biochem. Pharmacol. 2009, 78, 686–692. [Google Scholar]
- Cohen, C.; Bergis, O.E.; Galli, F.; Lochead, A.W.; Jegham, S.; Biton, B.; Leonardon, J.; Avenet, P.; Sgard, F.; Besnard, F.; et al. Ssr591813, a novel selective and partial alpha4beta2 nicotinic receptor agonist with potential as an aid to smoking cessation. J. Pharmacol. Exp. Ther. 2003, 306, 407–420. [Google Scholar] [CrossRef]
- Mao, D.; Gallagher, K.; McGehee, D.S. Mechanisms of Nicotine-Induced Excitatory Synaptic Plasticity in the VTA. In Proceedings of the 17th Annual Meeting of Society for Research on Nicotine and Tobacco, Toronto, Canada, Febuary 16–19, 2011; Society for Research on Nicotine and Tobacco: Madison, WI, USA, 2011. [Google Scholar]
- Hamid, S.; Dawe, G.S.; Gray, J.A.; Stephenson, J.D. Nicotine induces long-lasting potentiation in the dentate gyrus of nicotine primed rats. Neurosci. Res. 1997, 29, 81–85. [Google Scholar] [CrossRef]
- Hudkins, M.; O’Neill, J.; Tobias, M.; Bartzokis, G.; London, E. Cigarette smoking and white matter microstructure. Psychopharmacology (Berl.) 2012, 221, 285–295. [Google Scholar]
- Paul, R.H.; Grieve, S.M.; Niaura, R.; David, S.P.; Laidlaw, D.H.; Cohen, R.; Sweet, L.; Taylor, G.; Clark, R.C.; Pogun, S.; et al. Chronic cigarette smoking and the microstructural integrity of white matter in healthy adults: A diffusion tensor imaging study. Nicotine Tob. Res. 2008, 10, 137–147. [Google Scholar]
- Jacobsen, L.K.; Picciotto, M.R.; Heath, C.J.; Frost, S.J.; Tsou, K.A.; Dwan, R.A.; Jackowski, M.P.; Constable, R.T.; Mencl, W.E. Prenatal and adolescent exposure to tobacco smoke modulates the development of white matter microstructure. J. Neurosci. 2007, 27, 13491–13498. [Google Scholar]
- Calderan, L.; Chiamulera, C.; Marzola, P.; Fabene, P.F.; Fumagalli, G.F.; Sbarbati, A. Sub-chronic nicotine-induced changes in regional cerebral blood volume and transversal relaxation time patterns in the rat: A magnetic resonance study. Neurosci. Lett. 2005, 377, 195–199. [Google Scholar] [CrossRef]
- Stolerman, I.P.; Fink, R.; Jarvik, M.E. Acute and chronic tolerance to nicotine measured by activity in rats. Psychopharmacologia 1973, 30, 329–342. [Google Scholar] [CrossRef]
- Brielmaier, J.; McDonald, C.; Smith, R. Immediate and long-term behavioral effects of a single nicotine injection in adolescent and adult rats. Neurotoxicol. Teratol. 2007, 29, 74–80. [Google Scholar]
- Iniquez, S.; Warren, B.; Parise, E.; Alcantara, L.; Schuh, B.; Maffeo, M.; Manojlovic, Z.; Bolanos-Guzman, C. Nicotine exposure during adolescence induces a depression-like state in adulthood. Neuropsychopharmacology 2008, 34, 1609–1624. [Google Scholar]
- Belluzzi, J.D.; Lee, A.G.; Oliff, H.S.; Leslie, F.M. Age-dependent effects of nicotine on locomotor activity and conditioned place preference in rats. Psychopharmacology (Berl.) 2004, 174, 389–395. [Google Scholar]
- Hahn, B.; Stolerman, I.P.; Shoaib, M. Kappa-opioid receptor modulation of nicotine-induced behaviour. Neuropharmacology 2000, 39, 2848–2855. [Google Scholar] [CrossRef]
- Li, Z.; DiFranza, J.; Wellman, R.; Kulkarni, P.; King, J. Imaging brain activation in nicotine-sensitized rats. Brain Res. 2008, 1199, 91–99. [Google Scholar] [CrossRef]
- Polesskaya, O.; Fryxell, K.; Merchant, A.; Locklear, L.; Ker, K.; McDonald, C.; Eppolito, A.; Smith, L.; Wheeler, T.; Smith, R. Nicotine causes age-dependent changes in gene expression in the adolescent female rat brain. Neurotoxicol. Teratol. 2007, 29, 126–140. [Google Scholar] [CrossRef]
- Shiffman, S. Tobacco “chippers”: Individual differences in tobacco dependence. Psychopharmacology (Berl.) 1989, 97, 539–547. [Google Scholar]
- Shiffman, S.; Paty, J.; Kassel, J.; Gnys, M.; Zettler-Segal, M. Smoking behavior and smoking history of tobacco chippers. Exp. Clin. Pschopharmacol. 1994, 2, 126–142. [Google Scholar]
- Gervais, A.; O’Loughlin, J.; Meshefedjian, G.; Bancej, C.; Tremblay, M. Milestones in the natural course of cigarette use onset in adolescents. Can. Med. Assoc. J. 2006, 175, 255–261. [Google Scholar] [CrossRef]
- Edwards, S.A.; Bondy, S.J.; Kowgier, M.; McDonald, P.W.; Cohen, J.E. Are occasional smokers a heterogeneous group? An exploratory study. Nicotine Tob. Res. 2010, 12, 1195–1202. [Google Scholar] [CrossRef]
- Dierker, L.; Donny, E.; Tiffany, S.; Colby, S.; Perinne, N.; Clayton, R.; Tobacco Etiology Research Network. The association between cigarette smoking and DSM-IV nicotine dependence among first year college students. Drug Alcohol Depend. 2007, 86, 106–114. [Google Scholar]
- Carpenter, M.; Garrett-Mayer, E.; Vitoc, C.; Cartmell, K.; Biggers, S.; Alberg, A. Adolescent nondaily smokers: Favorable views of tobacco yet receptive to cessation. Nicotine Tob. Res. 2009, 11, 348–355. [Google Scholar] [CrossRef]
- Peterson, A.V., Jr.; Kealey, K.A.; Mann, S.L.; Marek, P.M.; Ludman, E.J.; Liu, J.; Bricker, J.B. Group-randomized trial of a proactive, personalized telephone counseling intervention for adolescent smoking cessation. J. Natl. Cancer Inst. 2009, 101, 1378–1392. [Google Scholar]
- DiFranza, J.; Ursprung, W.; Carlson, A. New insights into the compulsion to use tobacco from a case series. J. Adolesc. 2010, 33, 209–214. [Google Scholar]
- Tong, E.; Ong, M.; Vittinghoff, E.; Perez-Stable, E. Nondaily smokers should be asked and advised to quit. Am. J. Prev. Med. 2006, 30, 23–30. [Google Scholar] [CrossRef]
- DiFranza, J.R.; Savageau, J.A.; Fletcher, K.; Pbert, L.; O’Loughlin, J.; McNeill, A.D.; Ockene, J.K.; Friedman, K.; Hazelton, J.; Wood, C.; et al. Susceptibility to nicotine dependence: The Development and Assessment of Nicotine Dependence in Youth-2 Study. Pediatrics 2007, 120, e974–e983. [Google Scholar] [CrossRef]
- DiFranza, J.; Riggs, N.; Pentz, M. Time to re-examine old definitions of nicotine dependence. Nicotine Tob. Res. 2008, 10, 1109–1111. [Google Scholar] [CrossRef]
- Daglish, M.R.; Weinstein, A.; Malizia, A.L.; Wilson, S.; Melichar, J.K.; Britten, S.; Brewer, C.; Lingford-Hughes, A.; Myles, J.S.; Grasby, P.; et al. Changes in regional cerebral blood flow elicited by craving memories in abstinent opiate-dependent subjects. Am. J. Psychiatry 2001, 158, 1680–1686. [Google Scholar] [CrossRef]
- Franklin, T.; Wang, Z.; Sciortino, W.; Harper, D.; Willhite, R.; Wang, J.; Detre, J.; O’Brien, C.; Childress, A. Brain Signature of Cigarette Cue-Induced Craving Is Greatest in Females. In Proceedings of the 12th Annual Meeting of the Society for Research on Nicotine and Tobacco, Orlando, FL, USA, Febuary 15–18, 2006; Society for Research on Nicotine and Tobacco: Madison, WI, USA, 2006. [Google Scholar]
- Brody, A.L.; Mandelkern, M.A.; London, E.D.; Childress, A.R.; Lee, G.S.; Bota, R.G.; Ho, M.L.; Saxena, S.; Baxter, L.R.; Madsen, D.; et al. Brain metabolic changes during cigarette craving. Arch. Gen. Psychiatry 2002, 59, 1162–1172. [Google Scholar] [CrossRef]
- Lim, H.K.; Pae, C.U.; Joo, R.A.; Yoo, S.S.; Choi, B.G.; Kim, D.J.; Lee, C.; Lee, C.U. fMRI investigation on cue-induced smoking craving. J. Psychiatr. Res. 2005, 39, 333–335. [Google Scholar] [CrossRef]
- Brody, A.; Mandelkern, M.; Olmstead, R.; Jou, J.; Tiongson, E.; Allen, V.; Scheibal, D.; London, E.D.; Tiffany, S.T.; Cohen, M. Neural Substrates of Resisting the Urge to Smoke. In Proceedings of the 12th Annual Meeting of the Society for Research on Nicotine and Tobacco, Orlando, FL, USA, Febuary 15–18, 2006; Society for Research on Nicotine and Tobacco: Madison, WI, USA, 2006. [Google Scholar]
- Wilson, S.J.; Sayette, M.A.; Delgado, M.R.; Fiez, J.A. Instructed smoking expectancy modulates cue-elicited neural activity: A preliminary study. Nicotine Tob. Res. 2005, 7, 637–645. [Google Scholar]
- David, S.P.; Munafo, M.R.; Johansen-Berg, H.; Smith, S.M.; Rogers, R.D.; Matthews, P.M.; Walton, R.T. Ventral striatum/nucleus accumbens activation to smoking-related pictorial cues in smokers and nonsmokers: A functional magnetic resonance imaging study. Biol. Psychiatry 2005, 58, 488–494. [Google Scholar]
- Rubinstein, M.; Luks, T.; Moscicki, A.; Dryden, W.; Rait, M.; Simpson, G. Smoking-cue induced brain activation in adolescent light smokers. J. Adolesc. Health 2010, 48, 7–12. [Google Scholar]
- Wagner, D.; Cin, S.; Sargent, J.; Kelley, W.; Heatherton, T. Spontaneous action representation in smokers when watching movie characters smoke. J. Neurosci. 2011, 31, 894–898. [Google Scholar] [CrossRef]
- Kilts, C.D.; Gross, R.E.; Ely, T.D.; Drexler, K.P. The neural correlates of cue-induced craving in cocaine-dependent women. Am. J. Psychiatry 2004, 161, 233–241. [Google Scholar]
- Garavan, H.; Pankiewicz, J.; Bloom, A.; Cho, J.K.; Sperry, L.; Ross, T.J.; Salmeron, B.J.; Risinger, R.; Kelley, D.; Stein, E.A. Cue-induced cocaine craving: Neuroanatomical specificity for drug users and drug stimuli. Am. J. Psychiatry 2000, 157, 1789–1798. [Google Scholar] [CrossRef]
- Maas, L.C.; Lukas, S.E.; Kaufman, M.J.; Weiss, R.D.; Daniels, S.L.; Rogers, V.W.; Kukes, T.J.; Renshaw, P.F. Functional magnetic resonance imaging of human brain activation during cue-induced cocaine craving. Am. J. Psychiatry 1998, 155, 124–126. [Google Scholar]
- Wexler, B.E.; Gottschalk, C.H.; Fulbright, R.K.; Prohovnik, I.; Lacadie, C.M.; Rounsaville, B.J.; Gore, J.C. Functional magnetic resonance imaging of cocaine craving. Am. J. Psychiatry 2001, 158, 86–95. [Google Scholar]
- Childress, A.R.; Mozley, P.D.; McElgin, W.; Fitzgerald, J.; Reivich, M.; O’Brien, C.P. Limbic activation during cue-induced cocaine craving. Am. J. Psychiatry 1999, 156, 11–18. [Google Scholar]
- Brody, A.L.; Mandelkern, M.A.; Lee, G.; Smith, E.; Sadeghi, M.; Saxena, S.; Jarvik, M.E.; London, E.D. Attenuation of cue-induced cigarette craving and anterior cingulate cortex activation in bupropion-treated smokers: A preliminary study. Psychiatry Res. 2004, 130, 269–281. [Google Scholar] [CrossRef]
- Tanabe, J.; Nyberg, E.; Martin, L.F.; Martin, J.; Cordes, D.; Kronberg, E.; Tregellas, J.R. Nicotine effects on default mode network during resting state. Psychopharmacology (Berl.) 2011, 216, 287–295. [Google Scholar]
- Domino, E.; Ni, L.; Xu, Y.; Koeppe, R.; Guthrie, S.; Zubieta, J.K. Regional cerebral blood flow and plasma nicotine after smoking tobacco cigarettes. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 319–327. [Google Scholar] [CrossRef]
- Zubieta, J.K.; Heitzeg, M.M.; Xu, Y.; Koeppe, R.A.; Ni, L.; Guthrie, S.; Domino, E.F. Regional cerebral blood flow responses to smoking in tobacco smokers after overnight abstinence. Am. J. Psychiatry 2005, 162, 567–577. [Google Scholar] [CrossRef]
- Wang, Z.; Faith, M.; Patterson, F.; Tang, K.; Kerrin, K.; Wileyto, E.; Detre, J.; Lerman, C. Neural substrates of abstinence-induced cigarette cravings in chronic smokers. J. Neurosci. 2007, 27, 14035–14040. [Google Scholar] [CrossRef]
- Pierce, R.C.; Kalivas, P.W. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res. Brain Res. Rev. 1997, 25, 192–216. [Google Scholar]
- Tassin, J.P.; Vezina, P.; Trovero, F.; Blanc, G.; Herve, D.; Glowinski, J. Cortico-subcortical interactions in behavioral sensitization: Differential effects of daily nicotine and morphine. Ann. N. Y. Acad. Sci. 1992, 654, 101–116. [Google Scholar] [CrossRef]
- Brody, A.; Olmstead, R.; London, E.; Farahi, J.; Meyer, J.; Grossman, P.; Lee, G.; Huang, J.; Hahn, E.; Mandelkern, M. Smoking-induced ventral striatum dopamine release. Am. J. Psychiatry 2004, 161, 1211–1218. [Google Scholar]
- Dawe, S.; Gerada, C.; Russell, M.A.; Gray, J.A. Nicotine intake in smokers increases following a single dose of haloperidol. Psychopharmacology (Berl.) 1995, 117, 110–115. [Google Scholar]
- Koob, G.F.; Caine, S.B.; Parsons, L.; Markou, A.; Weiss, F. Opponent process model and psychostimulant addiction. Pharmacol. Biochem. Behav. 1997, 57, 513–521. [Google Scholar] [CrossRef]
- Koob, G.F.; Le Moal, M. Drug abuse: Hedonic homeostatic dysregulation. Science 1997, 278, 52–58. [Google Scholar] [CrossRef]
- Cole, D.M.; Beckmann, C.F.; Long, C.J.; Matthews, P.M.; Durcan, M.J.; Beaver, J.D. Nicotine replacement in abstinent smokers improves cognitive withdrawal symptoms with modulation of resting brain network dynamics. Neuroimage 2010, 52, 590–599. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Ryde, I.T.; Tate, C.A.; Seidler, F.J. Lasting effects of nicotine treatment and withdrawal on serotonergic systems and cell signaling in rat brain regions: Separate or sequential exposure during fetal development and adulthood. Brain Res. Bull. 2007, 73, 259–272. [Google Scholar] [CrossRef]
- Trauth, J.A.; Seidler, F.J.; McCook, E.C.; Slotkin, T.A. Adolescent nicotine exposure causes persistent upregulation of nicotinic cholinergic receptors in rat brain regions. Brain Res. 1999, 851, 9–19. [Google Scholar] [CrossRef]
- Trauth, J.A.; Seidler, F.J.; Slotkin, T.A. Persistent and delayed behavioral changes after nicotine treatment in adolescent rats. Brain Res. 2000, 880, 167–172. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Ryde, I.T.; Seidler, F.J. Separate or sequential exposure to nicotine prenatally and in adulthood: Persistent effects on acetylcholine systems in rat brain regions. Brain Res. Bull. 2007, 74, 91–103. [Google Scholar] [CrossRef]
- Kenny, P.J.; Markou, A. Nicotine self-administration acutely activates brain reward systems and induces a long-lasting increase in reward sensitivity. Neuropsychopharmacology 2006, 31, 1203–1211. [Google Scholar]
- Swanson, C.; Baker, D.; Carson, D.; Worley, P.; Kalivas, P. Repeated cocaine administration attenuates group I metabotproic glutamate receptor-mediated glutamate release and behavioral activation: A potential role for homer. J. Neurosci. 2001, 21, 9043–9052. [Google Scholar]
- Bowers, M.; McFarland, K.; Lake, R.; Peterson, Y.; Lapish, C.; Gregory, M.; Lanier, S.; Kalivas, P. Activator of G protein signaling 3: A gatekeeper of cocaine sensitization and drug seeking. Neuron 2004, 42, 269–281. [Google Scholar]
- Mowla, S.; Farhadi, H.; Pareek, S.; Atwal, J.; Morris, S.; Seidah, N.; Murphy, R. Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J. Biol. Chem. 2001, 276, 12660–12666. [Google Scholar]
- Theonen, H. Neurotrophins and neuronal plasticity. Science 1995, 270, 593–598. [Google Scholar]
- Schabitz, W.; Steigleder, T.; Cooper-Kuhn, C.; Schwab, S.; Sommer, C.; Schneider, A.; Kuhn, G. Intravenous brain-derived neurotrophic factor enhances post-stroke sensorimotor recovery and stimulates neurogenesis. Stroke 2007, 38, 2165–2172. [Google Scholar]
- Beuten, J.; Ma, J.; Payne, T.; Dupont, R.; Quezada, P.; Huang, W.; Crews, K.; Li, M. Significant association of bdnf haplotypes in European-American male smokers but not in European-American female or African-American smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 139, 73–80. [Google Scholar]
- Lang, U.E.; Sander, T.; Lohoff, F.; Hellweg, R.; Bajbouj, M.; Winterer, G.; Gallinat, J. Association of the met66 allele of brain-derived neurotrophic factor (BDNF) with smoking. Psychopharmacology (Berl.) 2007, 190, 433–439. [Google Scholar]
- Flatscher-Bader, T.; Zuvela, N.; Landis, N.; Wilce, P. Smoking and alcoholism target genes associated with plasticity and glutamate transmission in the human ventral tegmental area. Hum. Mol. Genet. 2008, 17, 38–51. [Google Scholar]
- Bhang, S.Y.; Choi, S.W.; Ahn, J.H. Changes in plasma brain-derived neurotrophic factor levels in smokers after smoking cessation. Neurosci. Lett. 2010, 468, 7–11. [Google Scholar]
- Kim, T.S.; Kim, D.J.; Lee, H.; Kim, Y.K. Increased plasma brain-derived neurotrophic factor levels in chronic smokers following unaided smoking cessation. Neurosci. Lett. 2007, 423, 53–57. [Google Scholar]
- Slotkin, T.; MacKillop, E.; Rudder, C.; Ryde, I.; Tate, C.; Seidler, F. Permanent, sex-selective effects of prenatal or adolescent nicotine exposure, separately or sequentially, in rat brain regions: Indices of cholinergic and serotonergic synaptic function, cell signaling, and neural cell number and size at six months of age. Neuropsychopharmacology 2007, 32, 1082–1097. [Google Scholar] [CrossRef]
- Uhl, G.R.; Liu, Q.-R.; Drgon, T.; Johnson, C.; Walther, D.; Rose, J.E.; David, S.P.; Niaura, R.; Lerman, C. Molecular genetics of successful smoking cessation: Convergent genome-wide association study results. Arch. Gen. Psychiatry 2008, 65, 683–693. [Google Scholar] [CrossRef]
- Garvey, A.; Bliss, R.; Hitchkock, J.; Heinold, J.; Rosner, B. Predictors of smoking relapse among self-quitters: A report from the normative aging study. Addict. Behav. 1992, 17, 367–377. [Google Scholar] [CrossRef]
- Shadel, W.; Martino, S.; Setodji, C.; Cervone, D.; Witkiewitz, K.; Beckjord, E.; Scharf, D.; Shih, R. Lapse-induced surges in craving influence relapse in adult smokers: An experimental investigation. Health Psychol. 2011, 30, 588–596. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Henningfield, J.E. Establishing a nicotine threshold for addiction. N. Engl. J. Med. 1994, 331, 123–125. [Google Scholar] [CrossRef]
- DiFranza, J.; Ursprung, W. The latency to the onset of nicotine withdrawal: A test of the sensitization-homeostasis theory. Addict. Behav. 2008, 33, 1148–1153. [Google Scholar] [CrossRef]
- Ursprung, S.; Morello, P.; Gershenson, B.; DiFranza, J. Development of a measure of the latency to needing a cigarette. J. Adolesc. Health 2010, 48, 338–343. [Google Scholar]
- Benowitz, N.L.; Kuyt, F.; Jacob, P. Circadian blood nicotine concentrations during cigarette smoking. Clin. Pharmacol. Ther. 1982, 32, 758–764. [Google Scholar] [CrossRef]
- Benowitz, N.; Jacob, P. Nicotine and cotinine elimination pharmacokinetics in smokers and nonsmokers. Clin. Pharmacol. Ther. 1993, 53, 316–323. [Google Scholar] [CrossRef]
- Kenny, P.; Markou, A. Neurobiology of the nicotine withdrawal syndrome. Pharmacol. Biochem. Behav. 2001, 70, 531–549. [Google Scholar] [CrossRef]
- Iyaniwura, T.T.; Wright, A.E.; Balfour, D.J. Evidence that mesoaccumbens dopamine and locomotor responses to nicotine in the rat are influenced by pretreatment dose and strain. Psychopharmacology (Berl.) 2001, 158, 73–79. [Google Scholar]
- Smith, K.M.; Mitchell, S.N.; Joseph, M.H. Effects of chronic and subchronic nicotine on tyrosine hydroxylase activity in noradrenergic and dopaminergic neurones in the rat brain. J. Neurochem. 1991, 57, 1750–1756. [Google Scholar]
- Miller, D.K.; Harrod, S.B.; Green, T.A.; Wong, M.Y.; Bardo, M.T.; Dwoskin, L.P. Lobeline attenuates locomotor stimulation induced by repeated nicotine administration in rats. Pharmacol. Biochem. Behav. 2003, 74, 279–286. [Google Scholar]
- Park, M.; Loughlin, S.; Leslie, F. Gestational nicotine-induced changes in adolescent neuronal activity. Brain Res. 2006, 1094, 119–126. [Google Scholar] [CrossRef]
- Perkins, K.A.; Gerlach, D.; Broge, M.; Grobe, J.; Sanders, M.; Fonte, C.; Vender, J.; Cherry, C.; Wilson, A. Dissociation of nicotine tolerance from tobacco dependence in humans. J. Pharmacol. Exp. Ther. 2001, 296, 849–856. [Google Scholar]
- Audrain-McGovern, J.; Rodriguez, D.; Tercyak, K.P.; Cuevas, J.; Rodgers, K.; Patterson, F. Identifying and characterizing adolescent smoking trajectories. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 2023–2034. [Google Scholar]
- Riggs, N.; Chou, C.-P.; Li, C.; Pentz, M. Adolescent to emerging adulthood smoking trajectories: When do smoking trajectories diverge, and do they predict early adulthood nicotine dependence? Nicotine Tob. Res. 2007, 9, 1147–1154. [Google Scholar] [CrossRef]
- DiFranza, J.; Wellman, R.; Ursprung, S.; Sabiston, C. The autonomy over smoking scale. Psychol. Addict. Behav. 2009, 23, 656–665. [Google Scholar] [CrossRef]
- DiFranza, J.; Ursprung, W.; Biller, L. The developmental sequence of tobacco withdrawal symptoms of wanting, craving and needing. Pharmacol. Biochem. Behav. 2012, 100, 494–497. [Google Scholar] [CrossRef]
- DiFranza, J.; Wellman, R.; Savageau, J. Does progression through the stages of physical addiction indicate increasing overall addiction to tobacco? Psychopharmacology (Berl.) 2012, 219, 815–822. [Google Scholar]
- DiFranza, J.; Sweet, M.; Savageau, J.; Ursprung, W. An evaluation of a clinical approach to staging tobacco addiction. J. Pediatr. 2011, 159, 999–1003. [Google Scholar] [CrossRef]
- DiFranza, J.; Wellman, R.; Mermelstein, R.; Pbert, L.; Klein, J.; Sargent, J.; Ahluwalia, J.; Lando, H.; Ossip, D.; Wilson, K.; et al. The natural history and diagnosis of nicotine addiction. Curr. Pediatr. Rev. 2011, 7, 88–96. [Google Scholar] [CrossRef]
- DiFranza, J.; Morello, P.; Gershenson, B. The retest reliability of nicotine dependence measures. Addict. Res. Theory 2011, 20, 55–63. [Google Scholar]
- Brody, A.L.; Mandelkern, M.A.; Jarvik, M.E.; Lee, G.S.; Smith, E.C.; Huang, J.C.; Bota, R.G.; Bartzokis, G.; London, E.D. Differences between smokers and nonsmokers in regional gray matter volumes and densities. Biol. Psychiatry 2004, 55, 77–84. [Google Scholar] [CrossRef]
- Zhang, X.; Salmeron, B.J.; Ross, T.J.; Geng, X.; Yang, Y.; Stein, E.A. Factors underlying prefrontal and insula structural alterations in smokers. Neuroimage 2011, 54, 42–48. [Google Scholar] [CrossRef]
- Gallinat, J.; Meisenzahl, E.; Jacobsen, L.K.; Kalus, P.; Bierbrauer, J.; Kienast, T.; Witthaus, H.; Leopold, K.; Seifert, F.; Schubert, F.; et al. Smoking and structural brain deficits: A volumetric mr investigation. Eur. J. Neurosci. 2006, 24, 1744–1750. [Google Scholar]
- Kuhn, S.; Schubert, F.; Gallinat, J. Reduced thickness of medial orbitofrontal cortex in smokers. Biol. Psychiatry 2010, 68, 1061–1065. [Google Scholar] [CrossRef]
- Gazdzinski, S.; Durazzo, T.C.; Studholme, C.; Song, E.; Banys, P.; Meyerhoff, D.J. Quantitative brain mri in alcohol dependence: Preliminary evidence for effects of concurrent chronic cigarette smoking on regional brain volumes. Alcohol. Clin. Exp. Res. 2005, 29, 1484–1495. [Google Scholar]
- Brown, R.W.; Kolb, B. Nicotine sensitization increases dendritic length and spine density in the nucleus accumbens and cingulate cortex. Brain Res. 2001, 899, 94–100. [Google Scholar] [CrossRef]
- Wellman, R.J.; DiFranza, J.R.; Savageau, J.A.; Godiwala, S.; Savageau, N.; Friedman, K.; Hazelton, J. The effect of abstinence on cigarette consumption upon the resumption of smoking. Addict. Behav. 2006, 31, 711–716. [Google Scholar] [CrossRef]
- Neuhaus, A.; Bajbouj, M.; Kienast, T.; Kalus, P.; von Haebler, D.; Winterer, G.; Gallinat, J. Persistent dysfunctional frontal lobe activation in former smokers. Psychopharmacology (Berl.) 2006, 186, 191–200. [Google Scholar]
- Jarvik, M.E. Further Observations on Nicotine as the Reinforcing Agent in Smoking. In Smoking Behavior: Motives and Incentives; Dunn, W.L., Jr., Ed.; V.H. Winston: Washington, DC, USA, 1973; pp. 33–49. [Google Scholar]
- McNeill, A.D.; West, R.; Jarvis, M.J.; Jackson, P.; Bryant, A.; Russell, M.A.H. Cigarette withdrawal symptoms in adolescent smokers. Psychopharmacology (Berl.) 1986, 90, 533–536. [Google Scholar]
- Goddard, E. Why children start smoking. Br. J. Addict. 1992, 87, 17–25. [Google Scholar] [CrossRef]
- Barker, D. Reasons for tobacco use and symptoms of nicotine withdrawal among adolescent and young adult tobacco users—United States, 1993. Morb. Mortal. Wkly. Rep. 1994, 43, 745–750. [Google Scholar]
- O’Loughlin, J.; Kishchuck, N.; DiFranza, J.; Tremblay, M.; Paradis, G. The hardest thing is the habit: A qualitative investigation of adolescent smokers’ experience of nicotine dependence. Nicotine Tob. Res. 2002, 4, 201–209. [Google Scholar] [CrossRef]
- Riedel, B.W.; Robinson, L.A.; Klesges, R.C.; McLain-Allen, B. Ethnic differences in smoking withdrawal effects among adolescents. Addict. Behav. 2003, 28, 129–140. [Google Scholar] [CrossRef]
- Strong, D.R.; Kahler, C.W.; Ramsey, S.E.; Abrantes, A.; Brown, R.A. Nicotine withdrawal among adolescents with acute psychopathology: An item response analysis. Nicotine Tob. Res. 2004, 6, 547–557. [Google Scholar] [CrossRef]
- Wellman, R.; DiFranza, J.; Wood, C. Tobacco chippers report diminished autonomy over tobacco use. Addict. Behav. 2006, 31, 717–721. [Google Scholar] [CrossRef]
- Panday, S.; Reddy, S.; Ruiter, R.; Bergstrom, E.; de Vries, H. Nicotine dependence and withdrawal symptoms among occasional smokers. J. Adolesc. Health 2007, 40, 144–150. [Google Scholar] [CrossRef]
- American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders, 4th ed; American Psychiatric Association: Washington, DC, USA, 1994.
- Stein, E.A.; Pankiewicz, J.; Harsch, H.H.; Cho, J.K.; Fuller, S.A.; Hoffmann, R.G.; Hawkins, M.; Rao, S.M.; Bandettini, P.A.; Bloom, A.S. Nicotine-induced limbic cortical activation in the human brain: A functional MRI study. Am. J. Psychiatry 1998, 155, 1009–1015. [Google Scholar]
- Domino, E.; Minoshima, S.; Guthrie, S.; Ohl, L.; Ni, L.; Koeppe, R.; Cross, D.; Zubieta, J. Effects of nicotine on regional cerebral glucose metabolism in awake resting tobacco smokers. Neuroscience 2000, 101, 277–282. [Google Scholar] [CrossRef]
- Rose, J.E.; Behm, F.M.; Westman, E.C.; Levin, E.D.; Stein, R.M.; Ripka, G.V. Mecamylamine combined with nicotine skin patch facilitates smoking cessation beyond nicotine patch treatment alone. Clin. Pharmacol. Ther. 1994, 56, 86–99. [Google Scholar] [CrossRef]
- Paterson, N.E.; Markou, A. Prolonged nicotine dependence associated with extended access to nicotine self-administration in rats. Psychopharmacology (Berl.) 2004, 173, 64–72. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
DiFranza, J.R.; Huang, W.; King, J. Neuroadaptation in Nicotine Addiction: Update on the Sensitization-Homeostasis Model. Brain Sci. 2012, 2, 523-552. https://doi.org/10.3390/brainsci2040523
DiFranza JR, Huang W, King J. Neuroadaptation in Nicotine Addiction: Update on the Sensitization-Homeostasis Model. Brain Sciences. 2012; 2(4):523-552. https://doi.org/10.3390/brainsci2040523
Chicago/Turabian StyleDiFranza, Joseph R., Wei Huang, and Jean King. 2012. "Neuroadaptation in Nicotine Addiction: Update on the Sensitization-Homeostasis Model" Brain Sciences 2, no. 4: 523-552. https://doi.org/10.3390/brainsci2040523
APA StyleDiFranza, J. R., Huang, W., & King, J. (2012). Neuroadaptation in Nicotine Addiction: Update on the Sensitization-Homeostasis Model. Brain Sciences, 2(4), 523-552. https://doi.org/10.3390/brainsci2040523